Article Text

Abstract
Objective Crohn's disease (CD) is caused by a complex interplay among genetic, microbial and environmental factors. ATG16L1 is an important genetic factor involved in innate immunity, including autophagy and phagocytosis of microbial components from the gut. We investigated the effect of inflammation on the composition of microbiota in the ileal mucosa of CD patients in relation to the ATG16L1 risk status.
Design Biopsies (n=35) were obtained from inflamed and non-inflamed regions of the terminal ileum of 11 CD patients homozygous for the ATG16L1 risk allele (ATG16L1-T300A) and 9 CD patients homozygous for the ATG16L1 protective allele (ATG16L1-T300). Biopsy DNA was extracted and the bacterial composition analysed by pyrosequencing. Intracellular survival rates of adherent-invasive Escherichia coli (AIEC) were analysed by determining colony forming units after exposure to monocytes isolated from healthy volunteers homozygous for the ATG16L1 risk or protective allele.
Results Inflamed ileal tissue from patients homozygous for the ATG16L1 risk allele contained increased numbers of Fusobacteriaceae, whereas inflamed ileal tissue of patients homozygous for the ATG16L1 protective allele showed decreased numbers of Bacteroidaceae and Enterobacteriaceae and increased Lachnospiraceae. The ATG16L1 allele did not affect the bacterial composition in the non-inflamed ileal tissue. Monocytes homozygous for the ATG16L1 risk allele showed impaired killing of AIEC under inflammatory conditions compared with those homozygous for the ATG16L1 protective allele.
Conclusions CD patients homozygous for the ATG16L1–T300A risk allele show impaired clearance of pathosymbionts in ileal inflammation indicating that ATG16L1 is essential for effective elimination of pathosymbionts upon inflammation.
- CROHN'S DISEASE
- IBD
- INTESTINAL MICROBIOLOGY
- GENOTYPE
- INTESTINAL BACTERIA
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Crohn's disease is an inflammatory disease of the gut caused by genetic, environmental and microbial factors.
The ATG16L1 gene has been identified as a susceptibility gene that is involved in innate immunity processes, including autophagy and phagocytosis of microbial components from the gut.
Crohn's disease is associated with a dysbiosis of microbiota.
What are the new findings?
Biopsies from inflamed parts of the ileum of patients with the ATG16L1 risk allele contained more Bacteroides, Fusobacteria and Escherichia coli and less Lachnospiraceae in comparison with patients with the ATG16L1 protective allele.
Activated monocytes isolated from volunteers with ATG16L1 risk allele showed impaired killing of adherent-invasive E. coli.
This study shows that the ATG16L1 risk phenotype appears under inflammatory conditions and causes bacterial dysbiosis at the mucosal level.
How might it impact on clinical practice in the foreseeable future?
Genotyping Crohn's disease patients for the ATG16L1 risk allele may help clinicians to adapt the prebiotic, pro-biotic or antibiotic therapy to control intestinal inflammation and correct the mucosal dysbiosis in these patients.
Introduction
The intestinal lumen is inhabited by a large number of microbes that aid in the digestion of dietary products.1 ,2 Healthy individuals are in symbiosis with the intestinal microbiota. Their intestinal immune system defends against pathogens3 and is tolerant towards resident commensal microbes. A disruption of the balance between the host organism and the intestinal microbiota triggers the activation of the intestinal immune system and initiates an inflammatory reaction, which is characteristic for intestinal disorders, such as IBDs.4 ,5
IBDs, mainly ulcerative colitis (UC) and Crohn's disease (CD), are inflammatory disorders that arise from a complex interplay between genetic susceptibility and environmental factors, where the mucosal immunity against commensal bacteria seems to play a crucial role.6 ,7 CD has a discontinuous inflammation that can occur in the entire GI tract, but is most typically located in the ileocolic region.7 ,8
Genome-wide association studies have created a comprehensive map of genomic susceptibility with over 160 loci that predispose for IBD,9 many of which are involved in antibacterial defence systems. However, very little is still known regarding the details of interactions between individual susceptibility variants and the specific microbial composition.10 ,11 Polymorphisms in the NOD2 gene have been linked to alterations in innate host immunity. In addition, polymorphisms in ATG16L1 and IRGM, two components involved in macro-autophagy, disturb the elimination of specific bacteria after internalisation through phagocytosis, linking disturbed autophagy to the pathogenesis of CD.12–15 Moreover, Paneth cells show an abnormal morphology in patients homozygous for the ATG16L1-T300A risk allele, which may affect the secretion of antibacterial peptides, such as defensins.16 ,17 Collectively, this may alter the microbiota composition and promote survival of intracellular bacteria in the underlying tissues, leading to chronic intestinal inflammation. Accumulating evidence support a tight link between phagocytosis and the autophagy machinery.18 ,19
Several studies have demonstrated that IBD patients have an altered microbiota composition compared with healthy individuals, showing a reduced diversity and an increase in mucosa-adherent bacteria.3 ,10 ,20 ,21 Gut microbiota undergo remodelling during the active phase of CD and differ between remission and relapse phases of disease, though it is unknown what drives this process.22 Compared with healthy controls, IBD patients have fewer bacteria with anti-inflammatory properties and/or more bacteria with pro-inflammatory properties.3 ,20 ,23 Faecalibacterium prausnitzii has anti-inflammatory properties and low numbers of it are associated with increased risk of postresection recurrence of ileal CD. In contrast, pro-inflammatory adherent-invasive Escherichia coli (AIEC) are more abundant in CD patients.3 Increased numbers of Bacteroides, fusobacteria and E. coli are associated with earlier relapse of CD in patients after ileocolectomy.3 ,24 We hypothesised that the ATG16L1 genotype may directly affect bacterial handling by the ileal mucosa in CD patients, favouring a pro-inflammatory state.
In the present study, we studied the interrelationship between the ATG16L1 genotype and the composition of microbiota in the inflamed and non-inflamed ileal mucosa of CD patients. In addition, monocytes from healthy volunteers were used to study the effect of the ATG16L1 genotype on the processing and killing of AIEC.
Material and methods
Tissue specimens
Ileal mucosal biopsies were obtained from CD patients at the University Medical Center Groningen, the Netherlands. All protocols for obtaining and studying human tissues were approved by the institution's Medical Ethical Committee UMCG. All patients gave written informed consent.
Intestinal biopsies were obtained from macroscopically inflamed and non-inflamed ileal mucosa from 9 CD patients homozygous for the ATG16L1 protective allele (ATG16L1-300) (P) and from 11 CD patients homozygous for the ATG16L1 risk allele (ATG16L1-T300A) (R).9 Biopsies were genotyped regarding NOD2 and IRGM genes as well. For paired analysis, 9 patients homozygous for risk allele provided biopsies from both inflamed and non-flamed regions and 6 patients homozygous for protective allele did as well. Biopsies were immediately snap-frozen in liquid nitrogen and stored at −80°C until further processing.
Patient data are described in online supplementary table S1.
DNA extraction
Total DNA was extracted from the biopsy samples using the QIAamp DNA mini kit (Qiagen, Germany) according to the manufacturer's instruction, with an additional bead beating cell lysis step using a Precellys 24 (Bertin Technologies, France) and glass beads at 5.5 ms−1 in 3 rounds of 1 min at room temperature. DNA was eluted from the columns by 2 sequential washes with 250 µL of low salt buffer.25 ,26
Pyrosequencing
Amplicon libraries for pyrosequencing of the 16S rDNA V1-V3 regions were generated using a barcoded forward primer consisting of the 454 Titanium platform A linker sequence, a key (barcode) that was unique for each sample, the 16S rRNA 534R primer sequence 5′-ATTACCGCGGCTGCTGG-3′, and a reverse primer consisting of a 9 : 1 mixture of two oligonucleotides, 5′-B-AGAGTTTGATCMTGGCTCAG-3′ and 5′-B-AGGGTTCGATTCTGGCTCAG-3′, where B represents the B linker followed by the 16S rRNA 8F and 8F-Bif primers, respectively.27 PCR amplifications were performed using 1× FastStart High Fidelity Reaction Buffer, 1.8 mM MgCl2, 1 mM dNTP solution, 5 U FastStart High Fidelity Blend Polymerase (Roche, USA), 0.2 µM reverse primer, 0.2 µM of the barcoded forward primer (unique for each sample) and 1 µL of template DNA. PCR was performed using the following cycle conditions: an initial denaturation at 94°C for 3 min, 25 cycles of denaturation at 94°C for 30 s, annealing at 51°C for 45 s and extension at 72°C for 5 min, and a final elongation step at 72°C for 10 min. Amplicons (20 µL) were purified using AMPure XP purification (Agencourt, USA) according to the manufacturer's instructions and eluted in 25 µL 1× low TE. Amplicon concentrations were determined by Quant-iT PicoGreen dsDNA reagent kit (Invitrogen, USA) using a Victor3 Multilabel Counter (Perkin Elmer, USA), and the quality was assessed on a Bioanalyzer 2100 (Agilent, USA). Amplicons were mixed in equimolar concentrations to ensure equal representation of each sample. A 454 sequencing run was performed on a GS FLX Titanium PicoTiterPlate with a GS FLX pyrosequencing system (Roche, USA).
Sequencing quality control
Pyrosequencing produced a total of 632 726 reads of 16S rRNA with an average of 12 000 reads per sample ranging from 5820 to 18 479 reads. Sequence analysis was performed using quantitative insights into microbial ecology28 with default parameters, including removing sequence artefacts using Denoiser29 and chimera removal with ChimeraSlayer, clustering via uclust30 at 97% similarity and then classified taxonomically using the Ribosomal Database Project (RDP) classifier31 retrained with Greengenes;32 in addition, for identification purposes down to the species level, using the phylogenetic software ARB as described by de Goffau et al.25
Monocyte isolation from human peripheral blood
Heparinised blood was obtained from 8 healthy volunteers homozygous for either the ATG16L1 protective or risk allele (4 volunteers each). Human peripheral blood mononuclear cells were isolated using Lymphoprep gradients (Axis-Shield PoC As, Norway). Monocytes were further purified using CD14 monoclonal antibodies conjugated to micro-beads according to the manufacturer's protocol (Miltenyi Biotec, Germany). The purity of monocytes was evaluated by fluorescent staining with CD14-FITC antibody (Miltenyi Biotec, Germany). Cell cultures of primary monocytes were performed in RPMI-1640 medium (PPA laboratories, Austria) supplemented with 10% (v/v) heat-inactivated fetal calf serum, penicillin (50 U/mL), streptomycin (50 µg/mL) and fungizone (5 µg/mL) at 37°C in a humidified atmosphere of 5% CO2. Monocytes were plated at 5×105 cells per mL on coverslips in 12-well plates with or without the presence of phorbol 12-myristate 13-acetate (PMA) (100 µM) (Sigma-Aldrich), TNFα and interleukin (IL)-1β to mimic inflammation.
Survival assay
The bacterial survival/killing was measured by the gentamicin protection assay as previously described.33 The numbers of colony forming units (CFUs) were determined and represent the AIEC that survived inside monocytes. To mimic inflammation, purified monocytes were activated overnight by PMA, TNFα and IL-1β.
RNA isolation and quantitative real-time PCR
Total RNA was extracted from tissue specimens with trizol (Sigma-Aldrich, The Netherlands), reverse transcribed and analysed for gene expression using real-time PCR (ABI PRISM 7700 sequence detector; Applied Biosystems, USA) as described before.34 TaqMan Gene Expression Assays for defensin 5 and 6 were obtained from Life Technologies (Bleiswijk, The Netherlands). CT values were normalised to the endogenous control (18S) and correlated inversely with initial mRNA levels. Primers and probes used are listed in online supplementary table S2. Quantitative PCR analysis for total 16S rRNA (representative of bacterial load) enumeration was performed.35 The ratio between 16S rRNA enumeration and 18S rRNA (representative of human tissue load) enumeration was calculated.
Statistical analysis
Principal component analysis (PCA) was performed to find clusters of similar groups of samples or species. All tests were performed with PASW Statistics 18 (SPSS, USA). As gut microbial species abundances are not normally distributed, non-parametric tests were used, as is described in the text. Differences in the CFUs and gene expressions were assessed by using the Mann–Whitney U test. All tests were two-tailed. p Values of 0.05 or lower were considered significant.
Results
There were no significant differences between bacterial burden of the patients homozygous for ATG16L1 protective allele and risk allele in inflamed and non-inflamed state (see online supplementary figure S1). The most abundant bacterial families in all the ileal mucosa samples were Bacteroideaceae (25%), Lachnospiraceae (18%), Enterobacteriaceae (9.4%), Ruminococcaceae (7.3%) and Fusobacteriaceae (6.4%) (see online supplementary figure S2).
Differences in the gut microbiota modification upon inflammation
Non-parametric analyses and PCA of the abundance of bacterial groups show that there are no significant differences in microbial composition in the non-inflamed ileal mucosa of CD patients homozygous for the ATG16L1 risk (R) or protective (P) allele. However, an unpaired analysis of the principal components and the individual microbial groups revealed significant differences in the composition of the gut microbiota upon inflammation in the 2 CD patient groups (figure 1). In the inflamed ileal mucosa, an upward shift with regard to principal component 2, which accounts for 14% of the variation within the data, was observed in patients homozygous for the ATG16L1 protective allele (PC2, p=0.01). PC2 was positively correlated with Lachnospiraceae and negatively with Bacteroidaceae (both p<0.001). Indeed, biopsies from the inflamed mucosa of CD patient homozygous for the ATG16L1 protective allele showed reduced levels of Bacteroidaceae compared with biopsies from the non-inflamed mucosa of these patients (p=0.008). Moreover, Lachnospiraceae were more common in the inflamed ileal mucosa of CD patients homozygous for the ATG16L1 protective allele compared with the inflamed ileal mucosa of CD patients homozygous for the risk allele (p=0.042).

Principal component analysis of the microbial composition on the family level. (A) First principal component (PC1) accounts for 54% of the variation in the data and second principal component (PC2) represents 14% of the variation. Samples from patients homozygous for the ATG16L1 risk allele are depicted by triangles (RI, risk allele inflamed ▴; RN, risk allele non-inflamed Δ) and samples from patients homozygous for the protective allele are depicted with circles (PN, protective allele non-inflamed ○; PI, protective allele inflamed •). (B) The main correlation with PC2 with relevant bacterial groups is tabulated. This table describes the correlation of four bacterial families related to the PC2. (C) The arrows in this figure indicate that the protective allele and inflamed mucosa group is positively correlated with PC2 (p=0.001) and contains relatively higher numbers of Lachnospiraceae and lower numbers of Bacteroidacea.
A paired analysis on patients from whom both inflamed and non-inflamed biopsies were available increased the resolution further with respect to detecting changes in the gut microbiota upon inflammation. For example, an increase in the number of Fusobacteriaceae is found upon inflammation in ileal biopsies of CD patients homozygous for the ATG16L1 risk allele (p=0.046). This increase was not observed in an unpaired analysis as patients who do not have any Fusobacteriaceae in non-inflamed ileal mucosa will also not have them in inflamed ileal mucosa. The analysis of paired inflamed and non-inflamed biopsies in the same patient confirmed the mucosal dysbiosis in CD patients homozygous for the ATG16L1 risk allele. Inflamed ileal mucosa from CD patients homozygous for the ATG16L1 protective allele showed relatively low numbers of Bacteroidaceae (p=0.028) and more Lachnospiraceae (p=0.046) in comparison with the non-inflamed ileal biopsies. In addition, inflamed mucosa form CD patients homozygous for the protective ATG16L1 allele had a lower score on PC3 (p=0.028), which is positively correlated with Enterobacteriaceae (p=0.016) and Fusobacteriaceae (p<0.001), indicating that these latter species are under-represented in the mucosa of these patients. In contrast, as described above, the inflamed ileal mucosa of CD patients homozygous for the ATG16L1 risk allele contained more Fusobacteriaceae than in the non-inflamed parts of their ileum (p=0.046).
A plot of the differences in microbial composition, which is a subtraction of non-inflamed from inflamed mucosa, in relation to the principal components 2 and 3 in CD patients homozygous for the ATG16L1 protective (P) or risk (R) allele is shown in figure 2A. This plot demonstrates the effect of inflammation on the mucosal microbiota of the 2 patient groups. CD patients with the protective allele are clustered together in the upper left part of figure 2A, indicating that upon inflammation the microbiota of the different patients of this group is modified in a similar pattern. In contrast, the differences in the paired samples of CD patients homozygous for the ATG16L1 risk allele are either close to zero in both dimensions (centre) or located on the right or near to the bottom of the plot, opposite to the samples from patients with the protective allele. In CD patients homozygous for the ATG16L1 protective allele, the localisation on figure 2A is due to a (relative) increase in the Lachnospiraceae numbers and a decrease in the numbers of Bacteroidaceae (PC2) and a decrease of Enterobacteriaceae and/or Fusobacteriaceae (PC3). In CD patients homozygous for the ATG16L1 risk allele, this is due to increased numbers of Fusobacteriaceae, a lack of changes or a combination of decreased numbers of Lachnospiraceae and increased numbers of Bacteroidaceae or Enterobacteriaceae. The direction and strength of the correlations are shown by arrows in figure 2B.

(A) Second principal component (PC2) that represents 14% of the variation in the data and the third principle component (PC3) representing 9% of the variation. Difference in subtraction of non-inflamed from inflamed state in 9 paired samples from patients homozygous for the ATG16L1 risk allele is depicted by ▴ and from 6 samples from patients homozygous for the ATG16L1 protective allele is depicted with •. The figure shows that the difference in paired protective samples is positively correlated with PC2 (p=0.001). (B) The direction and strength of the correlation with Fusobacteriaceae, Enterobacteriaceae and Bacteroidaceae are shown in arrows. (C) Shows the difference between percentage of Bacteroidaceae+Enterobacteriaceae+Fusobacteriaceae in inflamed and non-inflamed tissue (p=0.01). The figure shows the significant decrease in numbers of opportunistic bacteria in Crohn's disease (CD) patients homozygous for the protective allele while the numbers stay approximately the same in CD patients homozygous for the ATG16L1 risk allele.
Figure 2C shows the difference between the sum of the abundance of Bacteroidaceae, Enterobacteriaceae and Fusobacteriaceae between paired (non-inflamed and inflamed) biopsies from both patient genotypes. It shows that inflammation leads to a significant decrease in these three bacterial groups (when considered as one group) in patients homozygous for the ATG16L1 protective allele. In contrast, these numbers remain approximately the same in patients with the ATG16L1 risk allele during inflammation. The modification of the gut microbiota, as expressed by this sum, is significantly different between the two patients groups (p=0.01) and suggests that patients homozygous for the ATG16L1 risk allele respond differently or fail to respond properly to mucosal microbiota upon inflammation.
Since ATG16L1 mutations and IRGM or NOD2 mutations are involved in bacterial clearance, we also studied the effect of carrying the IRGM rs13361189 and NOD2 rs2066844 (R702W) and rs2066845 (G908R) variants. There were no patients homozygous for these IRGM and NOD2 risk alleles and patients heterozygote for these risk alleles showed no differences in our PCA.
Survival assay for AIEC
The survival of the AIEC, established as CFU after exposure to PMA-activated primary monocytes, was significantly higher using monocytes homozygous for the ATG16L1 risk allele compared with monocytes homozygous for the ATG16L1 protective allele, with an average of 173 vs 50 CFU, respectively (p<0.05) (figure 3A). No significant difference in CFU was observed when monocytes were not activated by PMA, indicating that differences in ATG16L1-dependent killing of AIEC only become apparent under inflammatory conditions. IL-1β stimulation of monocytes resulted in an increased numbers of CFUs in both groups of monocytes (p<0.05); however, this increase for monocytes homozygous for the ATG16L1 risk allele tended to be higher (average of 220 CFUs) than the one for monocytes homozygous for the ATG16L1 protective allele (average of 122 CFUs) (figure 3B). There were no significant differences between the survival rate of AIEC in the two types of monocytes with and without TNFα stimulation. However, as observed for IL-1β, CFU numbers using monocytes homozygous for the risk allele tended to be higher with an average of 96 vs 41 surviving AIEC in monocytes homozygous for the protective allele (figure 3C).

Differences in colony forming units (CFUs) of adherent-invasive Escherichia coli (AIEC) obtained from monocytes isolated from healthy volunteers homozygous for the ATG16L1 protective (P) or the risk allele (R) with and without simulation of inflammation by phorbol 12-myristate 13-acetate (PMA), IL-1β and TNFα. (A) Shows that there are no significant differences in CFU in volunteers with the protective allele both in stimulated conditions with PMA and un-stimulated conditions. In contrast, significantly higher numbers of survived AIEC bacteria were observed in stimulated monocytes with PMA or IL-1β from volunteers homozygous for the ATG16L1 risk allele compared with the un-stimulated situation (A and B) (p<0.05). (C) Shows the same trend for TNFα; however, the increase in CFU is not significant.
Differences in inflammation-related gene expression
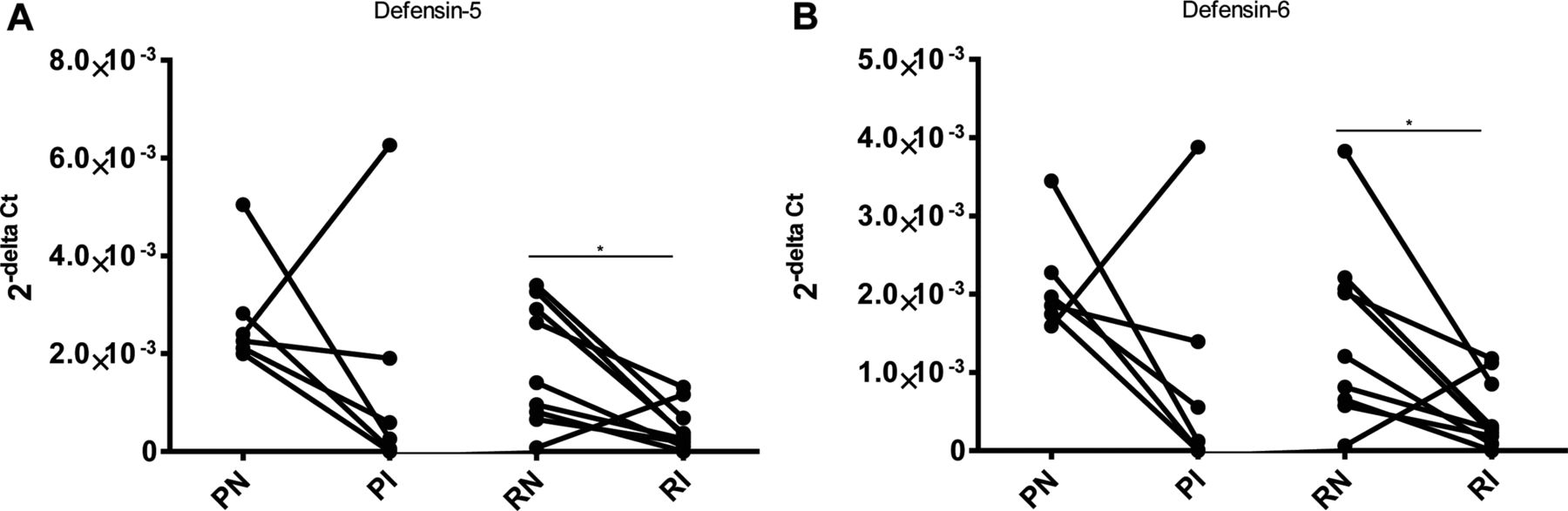
The NFκB mediated inducible nitric oxide synthase was equally increased in the inflamed ileum of both ATG16L1 genotypes36 (see online supplementary figure S3A). Except for 1 patient homozygous for the ATG16L1 protective allele, the defensin 5 and 6 gene expression (Paneth cell function) decreased upon inflammation in all other patients (p<0.05 for risk allele group) (figure 4A,B).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Differences in defencin 5 and 6 gene expression between inflamed and non-inflamed paired samples of patients homozygous for ATG16L1 risk (R) and protective allele (P). (A and B) Show that there is no significant difference in the gene expressions between inflamed biopsies of both group of patients (PI and RI).
The expression of various other cytokines, COX2, IL-1β (related to pro-inflammatory type 1 macrophages) and IL-10, MRC1 and TGF-β gene (related to tissue repair type 2 macrophages), was not significantly different in the inflamed ileum of both patients groups (see online supplementary figure S3).
Discussion
In this study, we show that CD patients homozygous for the ATG16L1 risk allele are unable to adequately clear pathosymbionts, such as Enterobacteriaceae, Bacteroidaceae and Fusobacteriaceae, during ileal inflammation in comparison with CD patients who are homozygous for the protective allele. Biopsies of inflamed terminal ileum from patients homozygous for the ATG16L1 protective allele contain markedly less pathosymbionts and have relatively more Lachnospiraceae than their counterparts homozygous for the risk allele. In contrast, no differences were observed between both groups when comparing the microbial composition of biopsies obtained from non-inflamed parts of the ileum. Using an in vitro inflammation model for isolated monocytes, we furthermore demonstrate the ATG16L1-T300A risk allele impairs xenophagy of invading opportunistic pathogens.
CD has been proposed to be the consequence of a immune response towards a variety of environmental inflammatory triggers in a genetic susceptible host.37 ,38 This can include the uncontrolled immune response against a variety of either pathogenic or non-pathogenic bacteria in the gut.4 ,5 Certain pathogenic bacterial species have been suspected as inflammatory triggers in CD. Specifically, the higher abundance of AIEC in pathogenesis of ileal CD and the role of potential pathogens such as Bacteroides fragilis and fusobacteria with regard to the recurrence of CD after ileocolonic resection have been described before.24 Additionally, it has been shown that the numbers of some non-pathogenic bacteria such as F. prausnitzii with beneficial anti-inflammatory effects on the epithelium significantly decrease in CD patients.3 These findings suggest a difference in the immune response between patients homozygous for ATG16L1 risk and protective allele with regard to killing invasive opportunistic pathogens, but that this is only observed in inflamed tissue. The higher abundance of Lachnospiraceae in biopsies from inflamed parts of CD patient's ileum homozygous for the protective allele could be due to the immune system's ability to distinguish non-pathogenic microbiota from pathosymbionts when harbouring this ATG16L1 gene variant.
A higher abundance of the three pathosymbiont groups during inflammation, namely, Enterobacteriaceae (mostly E. coli), Bacteroidaceae (mostly B. fragilis group) and Fusobacteriaceae, in inflamed tissue of the terminal ileum of patients with the risk allele, in comparison with those with the protective allele, is indicative of the impairment of the immune system of these patients to clear such bacterial groups. This could be a result of an impaired autophagy/xenophagy process. It remains elusive whether the invasion of the intestinal epithelial layer of patients homozygous for the risk allele by Enterobacteriaceae, Bacteroidaceae and Fusobacteriaceae is a cause or consequence of inflammation.
Our findings concerning the inability of the immune system of patients homozygous for the ATG16L1 risk allele to properly handle invading bacteria are supported by the results of the killing/survival assay. The higher numbers of bacterial CFU show the inability of monocytes homozygous for the ATG16L1 risk allele to effectively process and remove AIEC cells upon inflammation. Interestingly, such a difference is not observed when monocytes were not exposed to PMA. PMA activates protein kinase C and triggers reactive oxygen species production, thereby causing oxidative stress and increased production of inflammatory cytokines, providing an in vitro model for inflammation.39 In addition, we show that exposure to either IL-1β or TNF-α enhanced the survival of AIEC in monocytes homozygous for the risk allele compared with monocytes homozygous for the protective allele, though these differences were not significant. This may imply that the enhanced survival of AIEC in ATG16L1-T300A homozygous monocytes, as observed after PMA stimulation, is a result of a synergistic effect of various cytokines. This finding is in line with the findings of Murthy et al40 which show the inability of knock-in mice harbouring the ATG16L1 risk variant in effective clearance of the ileal pathogen Yersinia enterocolitica. The increased survival of AIEC in stimulated ATG16L1-T300A homozygous monocytes is in line with the increased numbers of potential pathogens like E. coli, B. fragilis and fusobacteria in inflamed mucosa of CD patients homozygous for the ATG16L1 risk allele. Furthermore, the fact that the ATG16L1 allele did not affect AIEC survival in non-stimulated monocytes is in agreement with the observation that no differences in the microbiota composition were found in non-inflamed tissue of the 2 patients groups. We did not find significant differences in the expression of genes related to pro-inflammatory signalling type 1 macrophages and tissue repair type 2 macrophages in the inflamed biopsies between two genotypes of patients or in biopsies from the non-inflamed ileum of those patients which could indicate that there are no differences in macrophages differentiation upon inflammation. However, the mechanism behind this inability and whether there is a difference in macrophages function upon activation needs to be further investigated.
Our hypotheses that the ATG16L1 risk allele impairs the autophagy process of pathogenic bacteria is in line with a previous study which showed that the ATG16L1 risk allele increases susceptibility to Helicobacter pylori infection.41 Moreover, in vitro studies revealed that the ATG16L1 risk allele incapacitates the autophagy progress against Salmonella in human epithelial cells42 and that siRNA knockdown of ATG16L1 impairs the autophagy process of AIEC in HeLa cells.43 In another study, the ATG16L1-T300A risk allele did not change the autophagy process against Salmonella typhimurium in mouse embryonic fibroblasts,44 which is also in agreement with our findings since differences only occur under inflammatory conditions.
Paneth cells produce different antimicrobial peptides, particularly the α-defensins 5 and 6. The expression of HD-5 and HD-6 in the paired (uninflamed vs inflamed) ileal biopsies from both genotypes was analysed. Both HD-5 and HD-6 expressions were strongly suppressed upon inflammation, but no significant differences were observed in HD-5 and HD-6 levels in non-inflamed ileum of the 2 genotypes or between the inflamed ileum of those two patients groups. These findings could indicate that the observed differences in mucosal bacterial composition are most probably not due to ATG16L1-related Paneth cell dysfunction.
Genotyping of CD patients for genes that are involved in bacterial recognition and autophagy could reduce postoperational recurrence of CD after ileocecal resection. Antibiotica propylaxis of recurrence has already been shown, but the use of more selective antibiotics to control the pathosymbionts could be even more beneficial, especially in patients homozygous for the ATG16L1 risk allele.45 Based on recent reports, one could also posit that small molecule inducers of selective autophagy could contribute to enhanced bacterial autophagy in patients homozygous for the ATG16L1 risk allele. However, additional studies are required to determine if the small molecules/FDA approved compounds possess both autophagy-dependent and autophagy-independent activity on immune pathways.46
ATG16L1 is a crucial factor in the autophagy pathway that is associated with the innate immune system.47 Mutation in this specific locus seems to affect the regulation of the immune response against the intestinal microbiota but only under conditions of inflammation. The ATG16L1 risk allele in CD patients is associated with a higher abundance of pathosymbionts, such as Enterobacteriaceae, Bacteroidaceae and Fusobacteriaceae, in the intestinal epithelial layer during inflammation. In contrast, patients with the protective allele have higher numbers of commensal bacteria such as Lachnospiraceae in their mucosal microbiota. These groups of bacteria may play a beneficial role in maintaining an anti-inflammatory balance since many of them are directly or indirectly involved in the production of butyrate, which stimulates the barrier function of the gut.48
In conclusion, this study shows that CD patients homozygous for the ATG16L1 risk allele display impaired clearance of pathosymbionts in ileal inflammation and that the killing of AIEC is impaired in activated monocytes homozygous for this risk allele, both indicating that ATG16L1 is essential for effective elimination of pathosymbionts upon inflammation.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figures
- Data supplement 2 - Online table 1
- Data supplement 3 - Online table 2
Footnotes
MSS and AR contributed equally.
Contributors MSS Designed and performed experiments, analysed the data and wrote the manuscript. AR Designed and performed experiments, wrote the manuscript. MCdG Analysed the data and performed statistical analysis. TB Performed experiments. RKW Recruited patients. JP Designed and performed experiments. KNF and HJMH Supervised the study, interpreted the data and critically revised the manuscript. GD Supervised the study, recruited patients, interpreted the data and critically revised the manuscript.
Funding MSS is supported by the Groningen University Institute for Drug Exploration (GUIDE). RKW is supported by a VIDI grant (016.136.308) from the Netherlands Organization for Scientific Research (NWO).
Competing interests None.
Ethics approval UMCG Ethical Committee, Protocol number 08/279.
Provenance and peer review Not commissioned; externally peer reviewed.