Article Text

Abstract
BACKGROUND Hereditary non-polyposis colorectal cancer (HNPCC) is one of the most common inherited disorders predisposing to cancer. The genes responsible for the disease have recently been cloned and characterised; their mutations induce a generalised genomic instability which is particularly evident at microsatellite loci (replication error (RER)+ phenotype).
AIMS To investigate how to select individuals and families in the general population who should be screened for constitutional mutations predisposing to colorectal cancer.
PATIENTS/METHODS Between 1984 and 1995, 1899 colorectal malignancies in 1831 patients were registered, and in 1721 of these (94%), family trees could be obtained. Patients and families were classified into five categories according to a more or less likely genetic basis: HNPCC; “suspected” HNPCC; juvenile cases; aspecific cancer aggregation; sporadic cases. In 18 families with HNPCC as well as in 18 with suspected HNPCC, microsatellite instability in tumour tissues and constitutional mutations of two DNA mismatch repair genes (MSH2 and MLH1) could be evaluated. RER status was studied with five markers (BAT40, D2S123,D18S57, D17S787, and BAT26) in paraffin embedded tissues. Germline mutations of MSH2 orMLH1 genes were assessed on DNA and RNA extracted from lymphomonocytic cells, using reverse transcription polymerase chain reaction, single strand conformation polymorphism analysis, and direct DNA sequencing.
RESULTS HNPCC represented 2.6% and suspected HNPCC 4.6% of all registered colorectal neoplasms. Eleven out of 18 HNPCC families (61%) showed microsatellite instability as opposed to four (of 18) suspected HNPCC (22%; p<0.02). Three germline mutations (two inMSH2 and one inMLH1 gene) were found in three different large HNPCC families, whereas no mutations were detected in suspected HNPCC.
CONCLUSIONS In this study of cancer genetic epidemiology, data from a tumour registry were analysed and this ultimately led to the identification and selection of families that should be tested for mutator gene mutations. With the use of a population based approach, the incidence of mutations was appreciably lower than previously reported and limited to families with full blown HNPCC. It is possible that in most families with a clinical spectrum of HNPCC (or suspected HNPCC) other DNA mismatch repair genes are involved in the pathogenesis of the disease.
- colorectal cancer
- hereditary non-polyposis colorectal cancer
- DNA repair genes
- mutation
- microsatellite loci
Abbreviations used in this paper
- HNPCC
- hereditary non-polyposis colorectal cancer
- RER
- replication errors
- PCR
- polymerase chain reaction
Statistics from Altmetric.com
- colorectal cancer
- hereditary non-polyposis colorectal cancer
- DNA repair genes
- mutation
- microsatellite loci
Colorectal malignancies probably represent the best example of the complex (and partially understood) interaction between environment and genetic background in the pathogenesis of a common tumour.1 In recent years, while relatively little has been learnt about exogenous agents, knowledge on the role of hereditary factors has been extended remarkably. Thus we now know that hereditary non-polyposis colorectal cancer (HNPCC or Lynch syndrome) is one of the most common hereditary conditions predisposing to cancer development; the disease is characterised by early onset colorectal cancer (before the age of 50 years), location of tumours in the right colon, and an increased risk of neoplasms of other organs, including endometrium, stomach, urothelium, small intestine, and ovary.2 The incidence of HNPCC is around 2–3% of all colorectal malignancies.3 ,4
The genes responsible for the HNPCC phenotype have recently been isolated and characterised. They are human homologues ofEscherichia coli (lower case) DNA mismatch repair genes, MSH2 andMLH1, 5 ,6 and account for the large majority of mutations found in families from various countries.7 In a few families, mutations of other mismatch repair genes (called PMS1,PMS2, GTBP/MSH6) have also been detected, although it seems that they do not have a major role in colorectal cancer predisposition. Mutations of these genes induce, in HNPCC tumours and a proportion of sporadic tumours, a generalised genomic instability, which is particularly evident at microsatellite loci (the so-called, replication errors, RER+ phenotype).8 ,9
The search for constitutional mutations of the DNA mismatch repair genes (also called “mutator genes”) is complex, expensive, and time consuming. It is unlikely therefore that such a test can be proposed as a screening procedure for colorectal cancer susceptibility in the general population. Thus an obvious question is how to select individuals and families in whom genetic testing for germline mismatch repair gene mutations would be more effective for early diagnosis and cancer prevention.
A specialised colorectal cancer registry was instituted in the local health care district in 1984. The main purposes of the registration were the study of familial occurrence of cancer and detection of families with HNPCC.10-12 The main objective of the present study is to evaluate how a detailed analysis of the registration data can be used to select individuals and families in the general population who should be screened for germline HNPCC mutations predisposing them to colorectal cancer.
Materials and methods
THE REGISTRY
Details of the general organisation of the specialised colorectal cancer registry have been described previously.13-16Health Care District 16 includes Modena and 10 surrounding communities made up of a total of 265 227 inhabitants (128 228 men and 136 939 women) at the 1991 census. Modena is in Northern Italy, 180 km south east of Milan. The area is highly industrialised (mainly cars, textiles, and pottery), entirely flat, almost exclusively urban, and with one of the highest incomes per person in Italy. The population density is 450/km2.
Registration of all colorectal malignancies in the district started in 1984; by the end of 1995, after a 12 year registration period, 1899 tumours in 1831 patients (953 men and 878 women) were detected, giving a crude incidence rate of 64.5 new cases/100 000/year in men and 55.2 in women. The corresponding figures for age adjusted (world population) incidence rates were 33.9 and 23.1 respectively. Histological verification was obtained in 97.6% of the tumours, with a mortality/incidence ratio of 0.70. Neoplasms of the large bowel were classified according to the International Classification of Diseases for Oncology (9th revision).17 Definitions such as “carcinoma in situ”, “neoplastic foci”, or “severe dysplasia” were not considered as cancer unless there was clear infiltration of the neoplastic tissue through the muscularis mucosae.
ANALYSIS OF PEDIGREES AND SUBGROUPING OF PATIENTS
The first page of the registration chart was reserved for personal data, general information on previous diseases, diet and life style, and an accurate genealogical tree, in which the main causes of morbidity and mortality of first degree relatives were recorded. During the 12 year period of registration, a total of 1721 “nuclear” pedigrees out of 1831 patients (94%) were collected and analysed. Information on close relatives could not be obtained for 110 registered individuals, owing to poor documentation; in our final analysis these were considered together with sporadic cases (table 1).
Main classes and relative incidence of colorectal carcinoma according to their possible or probable genetic origin. The data refer to the 12 year registration period 1984–95
Nuclear pedigrees were then classified and subdivided according to the presence of less than two, two, three, four, or more clinical criteria all indicative of an increased susceptibility to hereditary colorectal cancer. As previously described,18 ,19 these criteria were “verticality” (parent and offspring affected by colorectal cancer or other tumours featuring HNPCC), “aggregation” (in the sibship of the proband, at least 50% of siblings affected by cancer), “early onset” (cancer developed before the age of 50, in the HNPCC spectrum), “localisation in the right colon” (caecum, ascending, transverse, and flexures), “multiple tumours” (both synchronous and metachronous), and “mucinous histological type” (presence of mucus in 50% or more of the tumour at histology). Nuclear pedigrees with two or more of these criteria were then extended to second and third degree relatives, with the underlying assumption that the more criteria present in a given pedigree, the higher the probability of discovering features of HNPCC in the extended family tree. As far as possible, the diagnosis of cancer among family members was verified by histological records, clinical charts, or death certificates; verification was almost complete for neoplasms developed between 1980 and 1995, 50% complete for cases developed between 1965 and 1979, and largely incomplete for the preceding years.
After this detailed procedure of definition, extension, and selection of family trees (and verification of cancer diagnosis), we came to a new classification of colorectal malignancies already analysed and discussed in a previous report.19 Briefly, patients (and families) were subgrouped into five main categories. If the minimum (“Amsterdam”) criteria proposed by the International Collaborative Group on HNPCC were met,20 the family was classified as HNPCC. When a given family did not meet these criteria completely but a strong clinical suspicion of HNPCC was nevertheless maintained, then it was defined as “suspected” HNPCC (or HNPCC-like).19 ,21 A third group was represented by families with one or more first degree relatives (besides the proband) affected by tumours at any site but without suspicion of HNPCC (usually because of the late age of onset of cancer or the different tumour spectrum). The fourth group included “juvenile cases”—that is, patients in whom colorectal cancer developed before the age of 50 (and without other features of HNPCC). Finally, the fifth group included patients older than 50 at diagnosis and whose families did not show other cancer cases among relatives (“sporadic cases”). Table 1shows how the 1831 patients with colorectal cancer registered between 1984 and 1995 were subdivided in these five groups.
We wondered whether this new classification and subgrouping of colorectal malignancies could be used in clinical practice for selecting, from the affected individuals, those families that should be studied for DNA replication errors and mutations of mismatch repair genes. All the families with HNPCC according to the criteria of the International Collaborative Group on HNPCC would be tested, plus a representative sample of the families labelled on clinical grounds as suspected HNPCC. In the HNPCC group, 18 probands could be evaluated, and in some families the tests could be executed in two or more affected individuals; the remaining 10 families of this group could not be tested either because of the absence of affected family members still alive (n = 6) or because the family refused to undergo genetic analysis for detection of mutations predisposing to cancer (n = 4). In the suspected HNPCC group, 18 probands were similarly assessed; although the Amsterdam criteria were not satisfied,20 all these families showed: (a) at least two consecutive generations affected by colorectal neoplasms or other tumours of the HNPCC spectrum, (b) at least one cancer developed before the age of 50, and (c) in the sibship of the proband, 50% or more of the siblings affected by cancer.19
GERMLINE MUTATIONS OFMSH2 ANDMLH1 GENES
The experimental procedure for assessing constitutional mutations of the major DNA mismatch repair genes has already been reported in detail.22 Briefly, a 20–40 ml whole blood sample was taken (in EDTA or heparin) for each subject tested. The samples were investigated by both reverse transcription polymerase chain reaction (PCR), to detect the presence of aberrant mRNAs, and single strand conformation polymorphism analysis on cDNA or genomic DNA, followed by direct DNA sequencing, to identify mutations.
ANALYSIS OF MICROSATELLITE INSTABILITY (RER)
Paraffin embedded tumour tissue and adjacent normal mucosa were microdissected with sterile scalpels into polypropylene tubes. Samples were incubated for two hours in xylene (room temperature) and pelleted for five minutes. After a wash in ethanol, samples were dried and 100 μl digestion buffer (1 M Tris/HCl, 0.5 M EDTA, 0.02% Tween 20, 100 μg/ml proteinase K) was added. After overnight incubation at 37°C, proteinase K was inactivated (80°C, 10 minutes) and the samples were pelleted again. The DNA of each sample was purified with an NaCl saturated solution, precipitated in ethanol, and dissolved in sterile water. A 50–100 ng portion of genomic DNA was used for PCR.
Five simple repeated sequences were amplified, using PCR, from tumour and normal tissue DNA; the markers used wereBAT40, D2S123,D18S57, D17S787, and BAT26. 23 PCR amplification was carried out in a volume of 25 μl containing 0.5 μM unlabelled primer, 200 μM dGTP, dTTP, dATP, 25 μM dCTP, 2.5 μCi [α-33P]dCTP, 1.5 mM MgCl2, 50 mM KCl, 10 mM Tris/HCl, pH 8.4, 1 U Taq polymerase, and 50–100 ng DNA as template. The reaction mixture was processed through 35–40 cycles consisting of one minute at 94°C, one minute at 55°C, and one minute at 72°C. PCR products were run in 6% denaturating polyacrylamide and visualised by autoradiography. Differences in the banding pattern between tumour and normal DNA indicated microsatellite instability; RER+ tumours were defined as those in which this instability was detected in at least two microsatellite loci.
The statistical significance of differences in the frequency of RER+ between HNPCC and suspected HNPCC was assessed by the χ2test.
Results
Among the various subgroups of patients with colorectal cancer registered between 1984 and 1995 (table 1), we focused on HNPCC (47 individuals in 28 families, 2.6% of the total) and suspected HNPCC (84 in 72 families, 4.6%). Figure 1 shows the fluctuations of the number of patients in these two classes of subjects by year of registration and the total number of registered patients (top curve). In the HNPCC group, several members of the same family were affected by cancer during the 12 year period, whereas this event was much more rare in suspected HNPCC. Figure 2A,B shows representative pedigrees of families with phenotypic expression of HNPCC or suspected HNPCC respectively. In one of them (fig 2A) a constitutional mutation in theMSH2 gene was detected in the proband (arrow, patient operated on for rectal cancer at age 43) and in his 18 year old asymptomatic daughter. Two other healthy individuals in this family were tested, but no germline mutations were found.
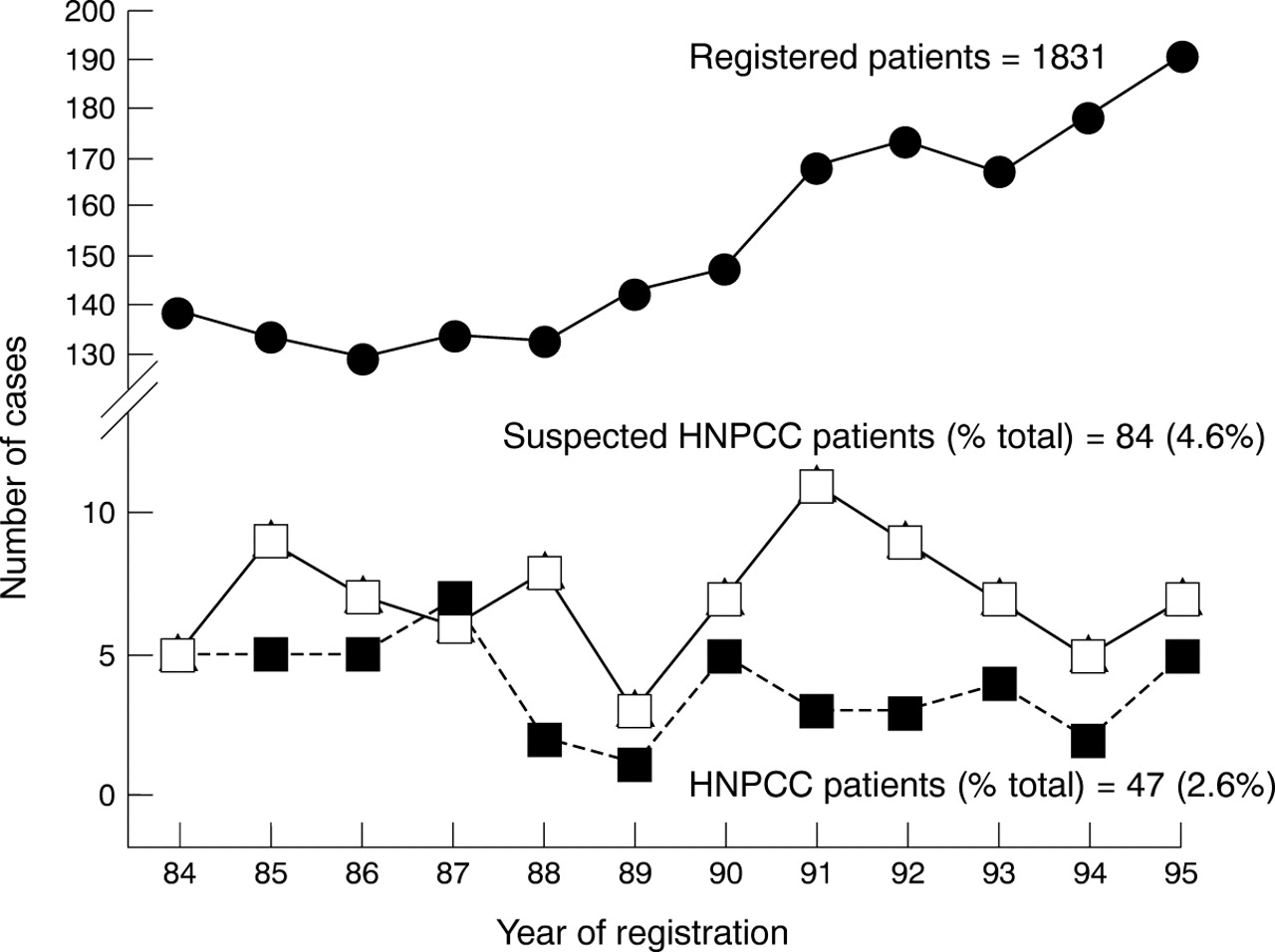
Patients with hereditary non-polyposis colorectal cancer (HNPCC) or suspected HNPCC observed in each year of registration (1984–1995). The upper part of the figure shows the total number of registered patients.


{kind=link}
{kind=link}
{kind=link}
(A) Representative pedigree of a family with full blown hereditary non-polyposis colorectal cancer (HNPCC) meeting the criteria of the International Collaborative Group on HNPCC (“Amsterdam Criteria”20). (B) Representative pedigree of a family with “suspected HNPCC”.
Table 2 shows the frequency of microsatellite instability and constitutional mutations of the major mutator genes (MSH2 and MLH1). As expected, most of the HNPCC families (61%) showed microsatellite instability at two or more loci (RER+ phenotype); in contrast, the RER+ status was less frequent among suspected families (22%, p<0.02). Only three germline mutations were detected in three different large families (all with a clinical spectrum of full blown HNPCC). Two of these mutations have previously been reported22: one was the already mentioned four base deletion (exon 7, codons 415–416) inMSH2 causing a frameshift and protein truncation; the second was an A to T substitution at the splice donor site of MSH2 intron 5. The third mutation was an A to C transversion within MLH1 codon 618 (exon 16) causing a lysine to tyrosine substitution.
Microsatellite instability and mutator gene mutations in HNPCC and suspected HNPCC families
Discussion
In this study, previously validated18 ,19 clinical criteria were used for stratifying and classifying all patients registered for colorectal tumours into different subgroups, according to a more or less likely genetic component. In the two subgroups who were at highest risk of genetically determined disease, microsatellite instability was assessed and constitutional mutations of the major DNA mismatch repair genes (MSH2 andMLH1) were searched for. Mutations were found only in families showing all clinical features of HNPCC, and their frequency (16.6%) was lower than that reported in previous investigations24 ,25; in contrast, no germline mutations were found in families with suspected HNPCC which, in addition, showed a much lower incidence of microsatellite instability.
In more recent years, biomolecular tests for detecting genetic predisposition to colorectal cancer have become available.26 ,27 As the techniques involved are complex, expensive, and time consuming, such tests cannot be used to screen the general population and have instead been applied to well defined series of colorectal cancer families, but not with a systematic population based approach. In line with the principles of genetic epidemiology,28 our approach began with a critical analysis of data from a cancer registry, and ultimately led to the identification and selection of subgroups of individuals and families who should be tested for mutator gene mutations. However, alternative strategies can also be used for the identification of HNPCC in the general population.
STRATEGIES FOR SELECTING INDIVIDUALS WITH GENETICALLY DETERMINED COLORECTAL TUMOURS
The large majority of neoplasms developing in HNPCC (about 90%) show microsatellite instability, whereas the RER+ phenotype is much less common in sporadic colorectal cancer (about 15%).29-31 These observations led de la Chapelle and collaborators to propose an attractive approach for the identification of HNPCC: if all colorectal malignancies developing in a given population could be assessed for the RER phenotype, then we should expect, from the literature, about 15% of positive tests; all positive cases should be screened for constitutional mutations ofMSH2 and MLH1genes.32 Although simple and straightforward, this approach needs considerable further laboratory work. Recent observations with this procedure seem to indicate an overall incidence of mutator gene mutations of 1–2% of all colorectal malignancies.32
Most of the known MSH2 andMLH1 mutations cause truncation of the proteins or absence of the transcripts.33 Recently, monoclonal antibodies have been developed which react with the proteins encoded by the two genes.34 ,35 Thus, by using standard immunohistochemistry, it is now possible to identify homozygous inactivation of either MSH2 orMLH1 by the absence of immunoreactive cells in tumour tissue. This can allow the screening of a large number of colorectal tumours with relatively simple immunohistochemical techniques, and the search for possible constitutional mutations of the major DNA mismatch repair genes in patients whose neoplasms are negative to immunostaining. As the technique cannot differentiate somatic from constitutional mutations of one allele (followed by inactivation of the other allele), it is likely that, as with RER testing, only a small proportion of the tumours lacking immunoreactive staining will represent HNPCC.
Among the various clinical approaches, a panel of experts recently proposed detailed guidelines (the “Bethesda Guidelines”) for the selection of colorectal tumours that should be tested for microsatellite instability and, ultimately, for the identification of HNPCC.36 These guidelines are mainly based on clinical criteria (age of onset of tumour, location in the large bowel, type of cancer, multiple malignancies), histopathological features (undifferentiated/cribriform pattern), and family history. According to these criteria, 15–20% of the total colorectal cancer burden in the population should be tested. This fraction is not far from that which can be obtained by adding together the HNPCC, suspected HNPCC, and juvenile cases in table 1, thus showing substantial similarities between the clinical criteria adopted in this study and those suggested by the National Cancer Institute.36
MICROSATELLITE INSTABILITY AND MUTATOR GENE MUTATION IN HNPCC
Several studies have indicated that the RER+ phenotype is usually found in most (70–100%) of the families showing clinical features of HNPCC.37-39 In line with these observations, 11 of 18 families (61%) assessed in this study showed microsatellite instability; moreover, mutator gene mutations were found only in RER+ families.
The proportion of HNPCC accounted for by MSH2and MLH1 mutations is still unsettled, whereas it is clear that the contribution of other genes implicated in the DNA mismatch repair mechanisms (PMS1, PMS2,GTBP/MSH6) is limited to a few families.40-42 In a comprehensive study of 48 kindreds with features of Lynch syndrome and microsatellite instability, Liuet al 43 reported mutator gene mutations in 70%. Partially at variance with these results, Tannergård et al 44 found germline mutations of MLH1 in eight of 39 Swedish HNPCC families; similar results (mutations ofMLH1 in 32% of the investigated families; no mutations in the MSH2 gene) were reported by Han et al 45 in Korea. Moreover, Weber et al 46identified constitutional mutations of MSH2or MLH1 genes in only 25% (eight of 32) of their American HNPCC families; the authors concluded that in most kindreds selected on clinical criteria, the molecular genetic basis of the disease remains to be determined. Our findings show an even lower incidence of mutations in typical HNPCC kindreds (table 2), and support the conclusion of Weber and coworkers,46 which are further substantiated by the population based approach. Lack of efficacy of our technique may be responsible for the low proportion of mutations being observed; however, this interpretation seems unlikely as our experimental procedure22 was similar to that of other investigations.42 ,44 ,46 Thus, germline mutations of the two major mismatch repair genes cannot account for all HNPCC families but only for a proportion of them. As most of these kindreds show microsatellite instability, it is likely that other, still unknown, mutator genes are involved; this implies that until the underlying genetic defects are determined, these families must be managed on a clinical basis.
MICROSATELLITE INSTABILITY AND MUTATOR GENE MUTATIONS IN SUSPECTED HNPCC
It is likely that the minimum requisites suggested for the research definition of HNPCC are too strict, and their rigid application may lead to many patients (and families) in whom colorectal cancer could have a genetic origin being missed.47 ,48This has led several researchers to gather together all kindreds that maintained a strong suspicion of being HNPCC even when the criteria for the clinical diagnosis were not entirely fulfilled. These were labelled “suspected”,49 “putative”,50 or “late onset”51 HNPCC. As expected, when biomolecular tests became available, microsatellite instability and mutator genes mutations were found in many of these families. In some studies,52 the mutation rate was rather low (12.5%), but in other investigations MSH2 orMLH1 mutations were detected in 24% of these families,45 and in a recent report Becket al 53 found mutations in 60%. However, this variation between studies is probably due mostly to variation in ascertainment.
Although we did not find mutator gene mutations in the 18 families investigated, the available body of evidence suggests that suspected HNPCC should receive the same consideration as classical Lynch syndrome, as Beck et al point out.53 The fact that tumours from patients with suspected HNPCC may show the RER+ phenotype is a further element that favours the systematic search of constitutional mutations of the major DNA mismatch repair genes in all these families. Moreover, there is evidence that genomic instability and mutator gene mutations occur in a definite fraction of young patients with colorectal cancer without other clinical features of HNPCC.54
In conclusion, our study showed one of the possible ways in which patients with colorectal cancer can be classified and stratified in various subgroups according to clinical criteria highly suggestive of a possible underlying genetic origin. Families showing features or suspicion of HNPCC should initially be tested in the RER assay; if positive, a search for constitutional mutations ofMSH2, MLH1, and possibly other DNA mismatch repair genes should be recommended. In practical terms this means analysing, by genetic tests, about 5% of all colorectal malignancies. Unfortunately only a few of them give positive results, at least at the present time.
Acknowledgments
This work was supported, in part, by grants from the Italian Ministry of the University (MURST, funds “60%” and “40%”), the Region Emilia-Romagna (Legge 1970, 15.03.1986), the Consiglio Nazionale delle Ricerche (CNR, ACRO project), the Associazione Italiana per la Ricerca sul Cancro (AIRC, special project “Hereditary Colorectal Carcinoma”) and the European Commission (contract BMH4- CTS6-0772).
Abbreviations used in this paper
- HNPCC
- hereditary non-polyposis colorectal cancer
- RER
- replication errors
- PCR
- polymerase chain reaction
References
Linked Articles
- Leading article