Article Text

Abstract
AIM Colorectal cancer has been described in association with hyperplastic polyposis but the mechanism underlying this observation is unknown. The aim of this study was to characterise foci of dysplasia developing in the polyps of subjects with hyperplastic polyposis on the basis of DNA microsatellite status and expression of the DNA mismatch repair proteins hMLH1, hMSH2, and hMSH6.
MATERIALS AND METHODS The material was derived from four patients with hyperplastic polyposis and between one and six synchronous colorectal cancers. Normal (four), hyperplastic (13), dysplastic (13), and malignant (11) samples were microdissected and a PCR based approach was used to identify mutations at 10 microsatellite loci, TGFβIIR, IGF2R, BAX, MSH3, and MSH6. Microsatellite instability-high (MSI-H) was diagnosed when 40% or more of the microsatellite loci showed mutational bandshifts. Serial sections were stained for hMLH1, hMSH2, and hMSH6.
RESULTS DNA microsatellite instability was found in 1/13 (8%) hyperplastic samples, in 7/13 (54%) dysplastic foci, and in 8/11 (73%) cancers. None of the MSI-low (MSI-L) samples (one hyperplastic, three dysplastic, two cancers) showed loss of hMLH1 expression. All four MSI-H dysplastic foci and six MSI-H cancers showed loss of hMLH1 expression. Loss of hMLH1 in MSI-H but not in MSI-L lesions showing dysplasia or cancer was significant (p< 0.001, Fisher's exact test). Loss of hMSH6 occurred in one MSI-H cancer and one MSS focus of dysplasia which also showed loss of hMLH1 staining.
CONCLUSION Neoplastic changes in hyperplastic polyposis may occur within a hyperplastic polyp. Neoplasia may be driven by DNA instability that is present to a low (MSI-L) or high (MSI-H) degree. MSI-H but not MSI-L dysplastic foci are associated with loss of hMLH1 expression. At least two mutator pathways drive neoplasia in hyperplastic polyposis. The role of the hyperplastic polyp in the histogenesis of sporadic DNA microsatellite unstable colorectal cancer should be examined.
- colon
- hyperplastic polyposis
- DNA mismatch repair
- microsatellite instability
Abbreviations used in this paper
- MSI-H
- MSI-L, microsatellite instability-high, -low
- MSS
- microsatellite stable
Statistics from Altmetric.com
The nature of the hyperplastic polyp remains unclear. It is accepted that small hyperplastic polyps found in the distal large bowel lack clinically significant malignant potential. An association with malignancy has been reported in subjects with hyperplastic polyposis, a condition that lacks a defined genetic basis but in which multiple large hyperplastic polyps are distributed throughout the colorectum.1-5 The precursor lesion could be a coexisting adenoma in such cases. Alternatively, larger hyperplastic polyps may carry an increased potential for dysplastic change. This fits with the finding of mixed polyps (appearing as a “collision” between classical hyperplastic polyp and adenoma) in cases of hyperplastic polyposis.1-5 In addition to hyperplastic polyps, adenomas and mixed polyps are serrated adenomas, lesions composed of epithelium that is simultaneously dysplastic and serrated.6 While accepted as an entity, the histological distinction between variant hyperplastic polyp and serrated adenoma is debated.7 ,8 Some have suggested that the polyps in hyperplastic polyposis are mainly serrated adenomas4(hence the term serrated adenomatous polyposis) but most reports of this condition describe a mixture of small and large hyperplastic polyps, mixed polyps, serrated adenomas, and traditional adenomas.1-3 ,5 ,7 ,9
Small classical hyperplastic polyps may show clonal genetic alterations.5 ,10-13 The term neoplastic is then warranted at the biological level, even though the lesion may be clinically innocuous.14 Nevertheless, newer molecular insights indicate that the rigid separation of serrated polyps into neoplastic and non-neoplastic classes may be artificial. A previous study of sporadic hyperplastic polyps, serrated adenomas, and mixed polyps derived from Japanese subjects indicated the possibility of a hyperplastic polyp-serrated adenoma pathway driven by a subtle form of genetic instability involving selected dinucleotide and complex DNA repeat sequences (in preference to mononucleotide).15
The aim of our study was to correlate the finding of DNA microsatellite instability with loss of DNA mismatch repair proteins in the hyperplastic and dysplastic components of serrated polyps from Western subjects with multiple large hyperplastic polyps. This was undertaken to determine whether or not large multiple hyperplastic polyps differ from their common small counterparts and to provide further insight into the molecular mechanisms driving the pathway of serrated neoplasia.
Materials and methods
The four patients included in the study had multiple large hyperplastic polyps and a personal but no family history of colorectal cancer. Patient No 1 was a woman aged 74 years with two cancers of the proximal colon. Both were contiguous with a large hyperplastic polyp but the smaller was also associated with a residual serrated adenoma. The right hemicolectomy specimen included four additional hyperplastic polyps that measured 8–15 mm. The case was selected for the study because in an initial work up both cancers were found to be microsatellite instability-high (MSI-H) and the large hyperplastic polyps contained foci of dysplasia that had lost hMLH1 expression (donated by AR). Patient No 2 was a 66 year old man with a cancer of the sigmoid colon, an early cancer limited to the head of a tubulovillous adenoma in the upper rectum, and 43 hyperplastic polyps ranging from 3 to 15 mm (donated by MS, DK, and DP). His appendix showed diffuse hyperplastic change. He underwent colonoscopy following sigmoid colectomy and a single adenoma was excised. A year later colonoscopic examination disclosed multiple polyps of 10–30 mm. Five were biopsied and shown to be hyperplastic polyps and one was a 13 mm mixed polyp combining a hyperplastic polyp with a tubular adenoma showing high grade dysplasia. The latter polyp as well as the cancers from the initial operative specimen were included in the present study. Patient No 3 was a 63 year old woman with a carcinoma of the ascending colon. Approximately 50 hyperplastic polyps measuring 2–25 mm were present throughout the subtotal colectomy specimen. Eight hyperplastic polyps with foci of dysplasia (mixed polyps) were included in the study. Patient No 4 (donated by DC) was a 66 year old woman with six synchronous cancers originating in the ascending colon (one), hepatic flexure (one), transverse colon (two), splenic flexure (one), and descending colon (one). Two were contiguous with serrated adenomas. There were 27 histologically confirmed polyps ranging from 5 to 12 mm. These included five hyperplastic polyps, five serrated adenomas, 13 mixed polyps, and four adenomas. The appendix showed diffuse hyperplasia with focal dysplasia.
As the aim of the study was to characterise foci of dysplasia within serrated polyps, tissue blocks were selected that included such lesions. The microdissected material included 11 cancers, 13 foci of dysplasia (figs 1A–3A), 13 hyperplastic epithelial samples, and normal mucosa from the four patients. Serial sections were cut for routine haematoxylin-eosin staining, microdissection for DNA extraction, and immunostaining for hMLH1, hMSH2, and hMSH6.

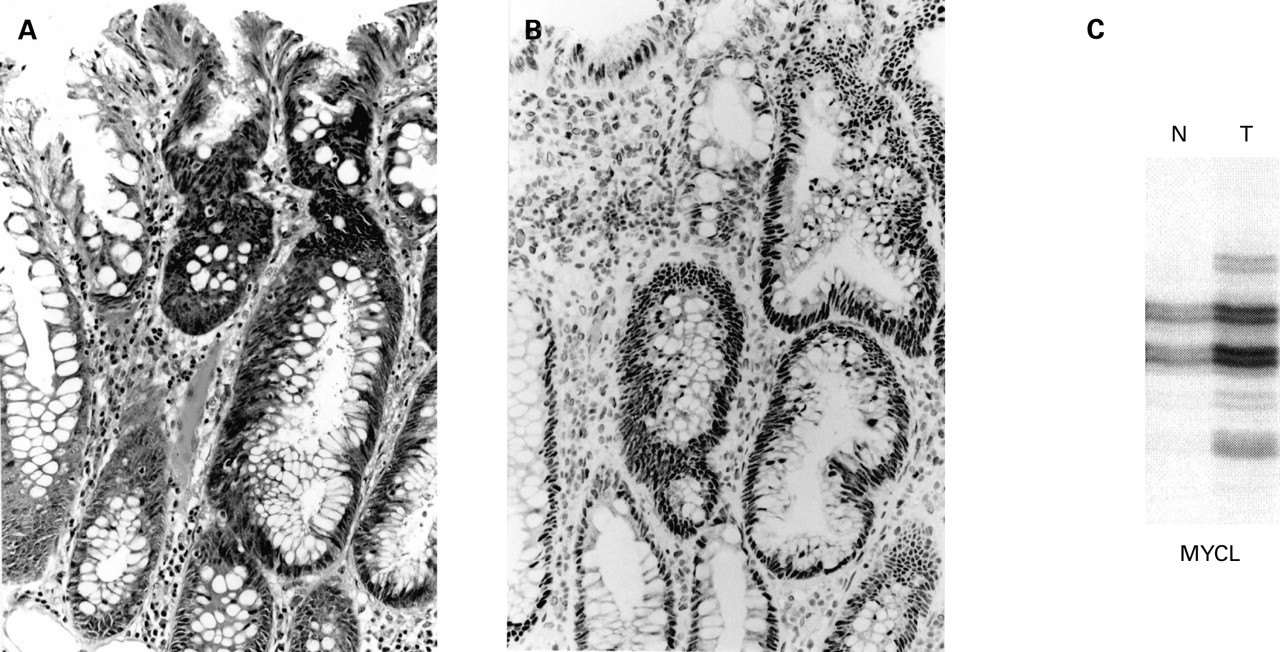
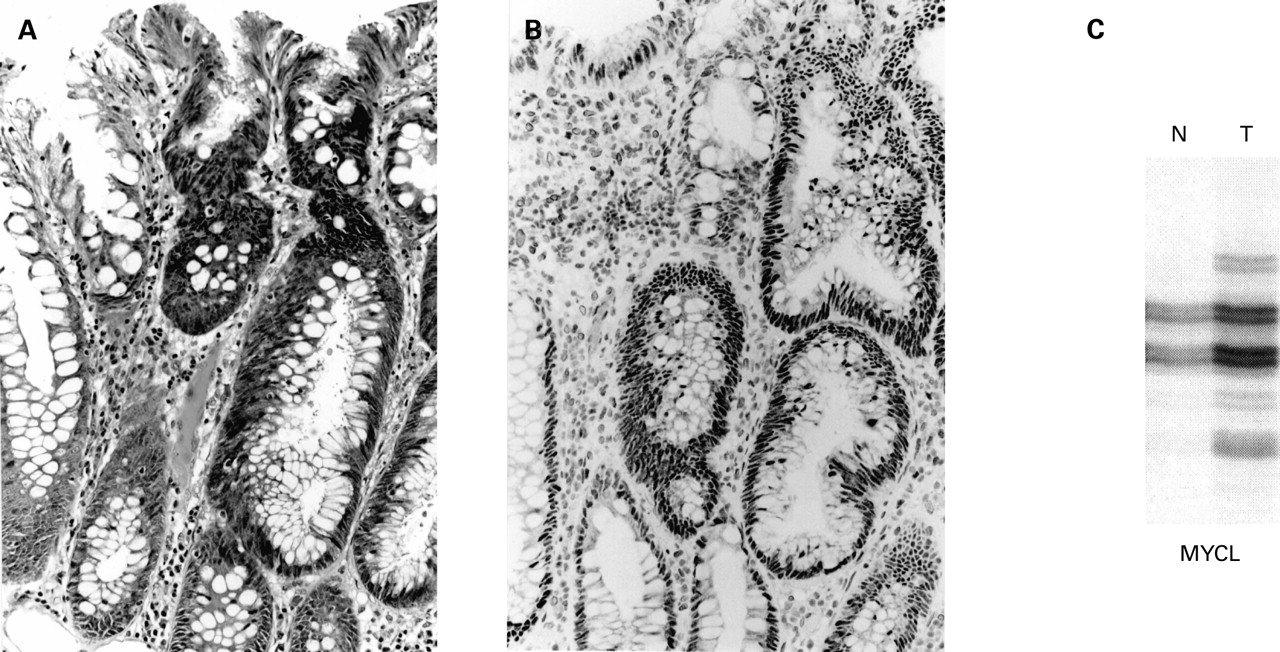
(A) Focus of dysplasia (right) arising in a hyperplastic polyp from patient No 1 (haematoxylin-eosin). (B) There is loss of nuclear staining for hMLH1 in the dysplastic area (ABC technique). (C) Bandshifts are shown for MYCL, BAT25, BAT26, and hMSH6. N, normal mucosa; T, tumour.
DNA PREPARATION AND ANALYSIS OF MICROSATELLITE INSTABILITY
DNA was extracted from microdissected sections of paraffin embedded archival sample as described previously.15 DNA from each pair of samples (normal mucosa and tumour) was analysed for MSI at 10 loci: MYCL,16 D2S123,17D5S346,18 D10S197,19 D18S69,19D18S58,20 BAT25,21 BAT26,21BAT40,21 and c-mybT22.22 The reaction volume for PCR was 10 μl of the DNA sample, 10 nM of each primer, 2 nM of each deoxynucleotide triphosphate, 1 μl of PCR buffer, 2.5 U of Taq polymerase (Boehringer Mannheim, Mannheim, Germany), and 1 μl of [33P] dATP. The PCR reaction was run as previously described.15 After amplification, PCR products were separated by electrophoresis in 5% denaturing polyacrylamide (19:1) gel and visualised by autoradiography. Tumours showing bandshifts at one or more microsatellite markers were termed MSI (figs 1C–3C). MSI status was subclassified as MSI-high (MSI-H) when bandshifts were seen at more than 40% of markers which showed informative PCR products and MSI-low (MSI-L) when the value was less than 40%. Lesions with no bandshifts were microsatellite stable (MSS). Scoring of MSI was undertaken independently by two observers (HI, JY).
MUTATION ANALYSIS OF REPEAT SEQUENCES OF TGFβR-II, IGF2R, BAX, hMSH3, AND hMSH6 GENES
The presence of mutation of the poly(A)10 tract of TGFβR-II,23 poly(G)8 tract of IGF2R,24poly(G) 8 tract of BAX,25 poly(A)8 tract of hMSH3,26 and poly(C)8 tract of hMSH626 was investigated by PCR and subsequent polyacrylamide gel electrophoresis. PCR was undertaken as for MSI testing. After amplification, PCR products were separated by electrophoresis in 5% or 8% denaturing polyacrylamide (19:1) gel according to the size of the products and visualized by autoradiography. The presence of bandshifts or an additional band was interpreted as mutation (figs 1C,2C).

(A) Junction of a hyperplastic polyp (right) and tubular adenoma (left) from patient No 2 (haematoxylin-eosin). (B) Loss of nuclear MLH1 is evident in the dysplastic tubules (ABC technique). (C) Bandshifts are shown for MYCL, BAT40, BAX, and hMSH3. N, normal mucosa; T, tumour.
IMMUNOHISTOCHEMISTRY
Paraffin sections (4 μm) were affixed to Superfrost Plus charged slides (Menzelgläser, Braunschweig, Germany) and dried overnight at 37°C. The sections were dewaxed and rehydrated in distilled water through descending graded alcohols. For hMLH1 and hMSH2, heat antigen retrieval was performed by autoclaving the sections in 0.001 M EDTA (pH 8.0) for 15 minutes. For hMSH6, microwave antigen retrieval (US patent No 5,244,787) was performed in 200 ml ChemMate HIER buffer pH 5.5–7.5 (Ventana, Tucson, Arizona, USA) for five minutes at 800 W followed by replenishment in H2O and a further five minutes at 800 W. After cooling, the sections were transferred to Tris buffered saline (TBS), pH 7.4. Endogenous peroxidase activity was quenched by incubating in 1% H2O2 and 0.1% NaN3 in TBS for 10 minutes. Following thorough washing in TBS, sections were immersed in 4% commercial non-fat skim milk powder in TBS for 15 minutes to inhibit non-specific antibody binding, before being transferred to a humidified chamber and covered with 10% normal (non-immune) goat serum for 30 minutes. Excess serum was decanted from the sections which were then incubated overnight at room temperature with primary monoclonal antibody to hMLH1 and hMSH2 and polyclonal rabbit antiserum to hMSH6. The monoclonal antibodies G168-15 (anti-hMLH1 diluted 1:75 in TBS) and G219-1129 (anti-hMSH2 diluted 1:100) were obtained from PharMingen (San Diego, California, USA). The polyclonal rabbit antiserum was raised against full length MSH6 protein, purified over a protein A column, and used in dilutions of 1:50 or 1:100.
Following incubations, sections were washed thoroughly in three changes of TBS for five minutes each. Sections were then incubated for 45 minutes with appropriate biotinylated secondary antibodies (Zymed Laboratories, San Francisco, California, USA) and then streptavidin-horseradish peroxidase conjugate (Zymed) for 15 minutes. Antigenic sites were revealed by incubating sections in 0.05% of 3,3′-diaminobenzidine in Tris saline with H2O2as substrate. After washing in gently running tap water, sections were lightly counterstained with haematoxylin, dehydrated through graded alcohols, cleared in xylene, and mounted with DePeX (figs 1B–3B).
Results
In the epithelium of normal mucosa, expression of hMLH1, hMSH2, and hMSH6 was confined to nuclei within the proliferative compartment of the crypts. This pattern was retained in hyperplastic crypts but two serrated polyps from patient No 4 included a small group of hyperplastic crypts showing loss of hMLH1 but no evidence of dysplasia. One of 13 (8%) hyperplastic polyp samples obtained by microdissection showed low levels of DNA microsatellite instability (MSI-L).
Dysplastic foci within the mixed polyps varied in size and morphology. Smaller foci comprised a few glands that were either “adenomatous” appearing or retained a serrated configuration with cytoplasmic eosinophilia but showed nuclear atypia. Larger dysplastic foci resembled traditional adenomas (tubular or tubulovillous), serrated adenomas, or intermediate forms. Of the 13 foci of dysplasia that were microdissected and an additional 21 that were not microdissected, 25/35 (71%) showed loss of hMLH1 (figs 1B, 2B), 2/14 (14%) loss of hMSH6, and none loss of hMSH2. When mismatch repair proteins were not lost in dysplastic foci, expression was sometimes increased (fig 3B) and present within a superficially located proliferative compartment.
(A) Focus of serrated dysplasia (right) arising in a hyperplastic polyp (haematoxylin-eosin). (B) There is accentuation of nuclear staining for hMLH1 (ABC technique). (C) The lesion shows instability at MYCL only. N, normal mucosa; T, tumour.
Loss of hMLH1 was seen in all four MSI-H foci of dysplasia (figs 1, 2; table 1) and in six MSI-H cancers from patient Nos 1 and 4. Both of the cancers from patient No 1 showed mutational bandshifts for hMSH6 but only one showed loss of expression of hMSH6. None of the three MSI-L foci of dysplasia (fig 3), the two MSI-L cancers (patient Nos 2 and 4), or the single MSI-L hyperplastic polyp showed loss of hMLH1 or hMSH6. The remaining three cancers (patient Nos 2, 3 and 4) were MSS. Loss of hMLH1 in MSI-H but not in MSI-L lesions showing dysplasia or cancer was significant (p<0.001, Fisher's exact test). Eight of 11 cancers were proximally located (5/6 MSI-H, 1/2 MSI-L, and 2/3 MSS). A summary of the results for all samples examined for both MSI status and expression of hMLH1, including mutated target genes, is shown in table 2.
Distribution of hMLH1 protein expression by DNA microsatellite status in dysplastic samples from mixed polyps and serrated adenomas
Results in 37 samples tested for both MSI status and hMLH1 expression
Discussion
In a selected sample of polyps from four subjects with multiple large hyperplastic polyps and synchronous carcinoma, changes in the form of loss of hMLH1 and occasionally hMSH6 expression and/or DNA microsatellite instability were mainly limited to foci of dysplasia and cancer. Only two of 13 microdissected dysplastic foci showed neither loss of hMLH1 expression nor DNA microsatellite instability. MSI-L was demonstrated in one of 13 (8%) samples of hyperplastic epithelium. Reports of dysplasia, adenoma, or carcinoma arising in hyperplastic polyps are uncommon but convincing. It has been suggested that hyperplastic polyps may progress to neoplasia on the basis that hyperplastic epithelium carries the same potential for neoplasia as normal large bowel epithelium.7 However, the current findings show that serrated neoplasia evolves through a specific pathway involving a defect in DNA mismatch repair. This would not be the most frequent pathway in the transition from normal to adenoma in the large bowel.
Genetic changes within typical hyperplastic polyps include the K-ras mutation as well as MSI-L but these are not consistent findings.5 ,10-12 Acquisition of the mild mutator phenotype appears to drive a particular pathway of neoplastic progression that does not involve loss of hMLH1 expression. MSI-L was found in three dysplastic foci in the current study. hMLH1 protein was present if not increased in these lesions (fig 3). The apparent increase may have been as compensation for loss of a different DNA mismatch repair mechanism or simply an expression of increased proliferation. MSI-L was demonstrated in contiguous foci of hyperplasia and serrated dysplasia in an earlier study of sporadic mixed polyps.15 The two MSI-L cancers in the current study retained hMLH1 expression. There are no reports in the literature of sporadic MSI-L cancers showing loss of hMLH1 expression.
Loss of hMLH1 expression may initiate a different pathway culminating in MSI-H. This occurred in patient No 1 in whom polyps were large but present in relatively small numbers. In patient No 4, four cancers and 15 dysplastic lesions (two adjacent to cancers) showed loss of hMLH1 expression. A marked variability of the hyperplastic polyposis phenotype was evident. Loss of hMLH1 was seen in four MSS foci of dysplasia from patient Nos 1 and 3. These were microscopic foci, and loss of the DNA mismatch repair protein may not have had time to establish significant levels of MSI. Alternatively, the microscopic subclones may simply have been cut out in the deeper section or diluted out by normal DNA. The four dysplastic lesions showing loss of hMLH1 protein and MSI-H were relatively large (figs 1, 2). The mechanism of loss of hMLH1 expression is likely to be due to hypermethylation of the promoter region rather than mutation.27 ,28 Such an epigenetic mechanism is supported by the high frequency of the finding, particularly evident in patient No 4.
Loss of hMSH6 expression (tested only in patient Nos 1–3) was limited to one MSI-H cancer showing mutation of hMSH6 (including contiguous residual serrated adenoma) and a small focus of MSS dysplasia that also showed loss of hMLH1. Mutational bandshifts in hMSH6 were found in an additional cancer (patient No 1) and a focus of dysplasia (patient No 2). As hMSH6 was expressed in the latter samples, these mutations were presumably monoallelic (epiphenomenal) and not evidence of a mutator mutation effect.26 However, the possibility of retained antigenicity with biallelic mutation (where one is missense) cannot be excluded.
The present findings support the view that the development of cancer in hyperplastic polyposis may begin within a hyperplastic polyp and may not depend on the presence of a coexisting traditional adenoma. DNA microsatellite instability occurs rarely in hyperplastic polyps but with relatively high frequency in dysplastic foci arising within the polyps of subjects with multiple large hyperplastic polyps. However, the molecular pathways to cancer appear to vary from case to case. Cancers developing in hyperplastic polyposis may be MSI-H, MSI-L, or MSS.5 ,11 Inactivation of hMLH1 may be a prerequisite for establishing the MSI-H pathway. The MSI-L pathway appears to implicate a separate and as yet unknown DNA repair mechanism. Neither hMSH3 nor hMSH6 is likely to be implicated as mutation of these putative secondary mutators26 was detected in MSI-H but not in MSI-L adenomas from HNPCC subjects (unpublished observations). Similarly, mutation of hMSH3 and hMSH6 and loss of hMSH6 expression were uncommon events in the current study. MSS cancers may either be underdiagnosed MSI-L cancers or could represent a third pathway involving coexisting traditional adenoma (fig 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Three possible pathways of neoplastic progression in hyperplastic polyposis. The adenoma (occurring either as a coincidental lesion or as a component of a mixed polyp) is shown as following the suppressor pathway. Hyperplastic polyps may evolve through the mild mutator/suppressor pathway or the high mutator pathway. Molecular characteristics of the three pathways are reported elsewhere.29 MMR, mismatch repair, MSI-H, MSI-L, microsatellite instability-high, -low; MSS, microsatellite stable.
The frequency of genetic instability in non-dysplastic regions of hyperplastic polyps from subjects with hyperplastic polyposis does not appear to be increased with respect to sporadic hyperplastic polyps or to approach that of serrated adenomas.15The non-dysplastic polyps in hyperplastic polyposis may therefore be large and dysplasia prone but otherwise indistinguishable from their smaller counterparts. Although this study sheds no light on the mechanism initiating the hyperplastic polyp or on the mismatch repair defect responsible for the MSI-L phenotype, the likely involvement of methylation in silencing hMLH127 ,28 suggests that methylation may also be implicated in earlier or initiating stages. Such an epigenetic mechanism would be consistent with polyp multiplicity and the tendency of hyperplastic polyps to regress spontaneously.30
In this study, MSI-H was found in 4/13 (31%) foci of dysplasia and in 6/11 (54%) cancers in subjects with hyperplastic polyposis (table 2). Additionally, 25/35 (71%) dysplastic lesions (from mixed hyperplastic polyps/adenomas or serrated adenomas) showed loss of hMLH1 (table 1). In an earlier study of sporadic serrated polyps, 5/29 (17%) mixed hyperplastic/adenomatous polyps or serrated adenomas were MSI-H.15 Dysplasia developing in the context of traditional colorectal adenomas may also be MSI-H. However, while this observation has been thoroughly documented in the case of HNPCC adenomas,31-34 there is an unexplained absence of MSI-H sporadic adenomas in the literature. One study found no MSI in sporadic adenomas35 but the same authors subsequently showed MSI in 3/49 sporadic adenomas containing high grade dysplasia.36In retrospect, one of these was a serrated adenoma and all three were MSI-L. Lothe et al described MSI in 7/70 sporadic adenomas but none showed high levels of MSI.13 A report documenting MSI in 20/73 proximal adenomas was not stringent in either excluding HNPCC adenomas or in distinguishing between MSI-L and MSI-H, and did not mention serrated adenomas.37
Conceivably MSI-H could be acquired at the point of transition from adenoma to carcinoma (analogous to TP53 in MSS cancers) but this is not supported by molecular data. The tumour suppressor gene APC plays a critical role in the initiation of colorectal adenoma. MSI-H cancers would therefore be expected to show APC alterations if the adenoma-carcinoma sequence applied. Yet an extremely low rate of APC mutation has been reported in MSI-H cancers38 ,39 and following full length sequencing of APC derived from MSI-H colorectal cell lines.40 We and others failed to show a single example of 5q LOH in 20 and nine MSI-H colorectal cancers, respectively.29 ,38
We suggested previously that MSI-L colorectal cancer might arise through neoplastic transformation of a hyperplastic polyp.15 It now seems likely that some, if not all, MSI-H cancers arise through a similar morphogenetic pathway. This would explain the infrequency of sporadic MSI-H adenomas and APC alterations in MSI-H cancers. The mechanism could be through transformation of a hyperplastic polyp or a serrated adenoma arising de novo. Given the numerous clinical, pathological, and molecular differences that have now been demonstrated between MSI positive and MSI negative colorectal cancers, it would perhaps be surprising if differences in early morphogenesis did not also exist.
Acknowledgments
We thank Brenda Mason for secretarial support and Clay Winterford for preparing the photomicrographs. This work was supported by a grant from the NIH/USA (No 1 UO1 CA74778–01; Collaborative Family Registry for Colorectal Cancer Studies).
Abbreviations used in this paper
- MSI-H
- MSI-L, microsatellite instability-high, -low
- MSS
- microsatellite stable