Article Text

Abstract
Background—Transforming growth factor α (TGF-α) knockout mice have increased susceptibility to dextran sodium sulphate (DSS) induced colitis.
Aim—To substantiate the findings that TGF-α is a key mediator of colonic mucosal protection and/or repair mechanisms by evaluating the susceptibility of mice overexpressing TGF-α to DSS induced colitis.
Methods—TGF-α overexpression was induced in transgenic mice by ZnSO4administration in drinking water (TG+). Three groups were used as controls: one transgenic group without ZnSO4 administration (TG−), and two non-transgenic littermate groups receiving ZnSO4 (Non-TG+) or only water (Non-TG−). Acute colitis was induced in all groups by administration of DSS (5%, w/v) in drinking water for six days ad libitum.
Results—About 35–39% of the entire colonic mucosa was destroyed in Non-TG−, Non-TG+, and TG− animals compared with 9% in TG+ mice. The crypt damage score was 18.7 (0.9), 18.2 (1.0), 18.9 (0.8), and 6.8 (1.5) (means (SEM)) in Non-TG−, Non-TG+, TG−, and TG+ mice respectively. Mucin and bromodeoxyuridine staining were markedly enhanced in colons of TG+ mice compared with controls, indicating increased mucosal protection and regeneration.
Conclusions—The significantly reduced susceptibility of mice overexpressing TGF-α to DSS further substantiates that endogenous TGF-α is a pivotal mediator of protection and/or healing mechanisms in the colon.
- transforming growth factor α
- epidermal growth factor
- dextran sodium sulphate
- colitis
- inflammatory bowel disease
- transgenic mice
Statistics from Altmetric.com
- transforming growth factor α
- epidermal growth factor
- dextran sodium sulphate
- colitis
- inflammatory bowel disease
- transgenic mice
Studies in laboratory animals have provided indirect evidence that among the epidermal growth factor (EGF) family of related peptides, both EGF and transforming growth factor α (TGF-α) play an important role in mucosal protection and healing of gastrointestinal lesions.1-7 The influence of other EGF family members—for example, heparin binding EGF, amphiregulin, and betacellulin—on mucosal protection and healing has not yet been established. EGF and TGF-α have a similar molecular mass, share 35% amino acid sequence homology, and are both initially synthesised as large transmembrane precursors.8-12 Furthermore, these mature growth factors, as well as their precursors, compete for binding to a common receptor and induce generally similar biological responses.1 ,13
In a rat model of colitis, we1 and others2have shown that intraperitoneal administration of EGF before induction of colitis with trinitrobenzenesulphonic acid/ethanol significantly reduces the degree of inflammation and severity of mucosal injury. We also observed a marked increase in TGF-α binding sites in the inflamed areas of an immune complex model of colitis in rabbits.14 Subsequent investigations in our laboratory showed enhanced expression of the mRNA for TGF-α but not for EGF within hours of the induction of colitis by trinitrobenzenesulphonic acid/ethanol in rats.15 We recently showed that TGF-α knockout mice as well as mice deficient in TGF-α (wa-1) are highly susceptible to dextran sodium sulphate (DSS) induced colitis when compared with genetic controls of knockouts and Balb/c mice.16 We have also shown that exogenous administration of TGF-α reduces the severity of acute DSS induced injury in wa-1 mice, an inbred strain deficient in TGF-α.16 These findings, together with the abundance of TGF-α in columnar epithelial and goblet cells throughout the colon of rats,17 suggested that TGF-α may be an important mediator of mucosal protection and/or repair mechanisms of the colonic mucosa.
One limitation of the use of null mutation (knockout) strains is the possibility that other genes may be overexpressed as a result of the engineered deletion. Thus, compensatory processes triggered by the introduced mutation may lead to erroneous conclusions about the molecule initially investigated.18-20 Furthermore, as some developmental pathways involve TGF-α expression,21-26 the phenotypes of adult knockout strains may be significantly altered from wild type. To evaluate the role of endogenous TGF-α as a mediator of protection and/or repair mechanisms in the colonic mucosa in the absence of these potentially complicating factors, we used a strain of mice that overexpresses biologically active TGF-α, developed by Sandgren et al.27 These investigators created the TGF-α transgenic line MT-TGF-α-mini 1745–8 (Tg (Mt-I, Tgf-α) Bril4c) by injecting into fertilised mouse eggs a transgene consisting of the metallothionein-I enhancer/promoter (MT), a TGF-α cDNA/genomic fusion sequence carrying introns 4 and 5, and a human growth hormone non-coding sequence containing the poly(A) adenylation site.27 The MT promoter is inducible with Zn2+and has been proven to be transcriptionally active in many adult tissues.27 TGF-α transgenic mice showed a uniform hyperplasia of adult liver, pancreas, stomach, small intestine, caecum, colon, and coagulation glands after induction with Zn2+. Thus these mice may be a better tool than knockout strains for studying the function of a single peptide, since MT allows a relatively efficient on/off switch mechanism of gene expression in adult mice, and effects from compensatory pathways created during embryological development can be excluded.
The aim of the present study was to substantiate our previous findings that TGF-α is a key mediator in repair/protection mechanisms of the colon by evaluating the susceptibility of transgenic mice overexpressing TGF-α to DSS induced colitis.
Materials and methods
ANIMALS
TGF-α transgenic mice and their non-transgenic littermates were bred at the School of Veterinary Medicine, University of Wisconsin, Madison. All mice (male) were five or six months old and were housed two per cage. They were grouped (10 per group) as transgenic mice with zinc administration (TG+), transgenic mice without zinc administration (TG−), non-transgenic control mice with zinc administration (Non-TG+), and non-transgenic control mice without zinc administration (Non-TG−). ZnSO4 (25 mM) was administered to the TG+ and Non-TG+ group in drinking water beginning two months before initiation of the study. This time point was selected to ensure that the transgenic mice had fully developed the effects of maximal induction of the transgene expression. Transgenic mice were identified by polymerase chain reaction (PCR) of tail DNA as described previously.27 Body weights at the start of the trial were not significantly different between the two transgenic groups (TG+, 31.4 (0.7) g and TG−, 33.0 (1.2) g) or the two control groups (Non-TG+, 36.7 (1.3) g and Non-TG−, 37.2 (2.2) g). However, there was a significant difference between transgenic and control animals (p<0.05).
INDUCTION OF ACUTE COLITIS
The two groups of TGF-α transgenic mice and the two groups of littermate controls (10 per group) were given 5% (w/v) DSS (molecular mass 40 kDa; ICN, Costa Mesa, California, USA) in distilled water ad libitum. A separate set of control animals corresponding to each of the four groups did not receive DSS (three per group). Mean DSS/water and distilled water consumption was noted per cage and day. Mice were weighed daily and visually inspected for diarrhoea and rectal bleeding. Animals were killed by carbon dioxide narcosis on day six. This period was selected because precipitous declines in body weight, profuse rectal bleeding, and progressive lethargy were noted by day five in all groups except TG+. In addition, three animals from the TG− group died at the beginning of day six. These animals have been excluded from the assessment of colitis and from statistical analysis.
GENERAL ASSESSMENT OF COLITIS
Colitis was assessed as previously described.16 The entire colon was dissected out and the length recorded. Colonic length of the controls (no DSS) was used as a reference. The colons were opened longitudinally, and gross blood content was assessed using a scale from 0 to 3+ (0, no blood; 1+, blood in up to one third of the colon; 2+, blood in up to two thirds of the colon; 3+, blood in more than two thirds of the entire colon). The entire colon as well as the stomach and parts of the small intestine were fixed in 10% zinc/formalin and embedded in paraffin, and transverse sections were cut and stained routinely with haematoxylin and eosin. Sequential high (1:40) power fields (31–64, depending on the length of the colon) of the entire colon were evaluated histologically, and the grade of injury was expressed as a percentage of each field for the entire colon. The severity of mucosal injury was graded as previously described16 from a scale of 0–III: grade 0, normal; grade I, distortion and/or destruction of the bottom third of the glands; grade II, erosions/destruction of bottom two thirds or loss of the entire glands with remaining surface epithelium; grade III, loss of the entire glands. Samples were evaluated independently by two investigators blinded to the strain of mice examined. Each grade of mucosal injury was determined for the entire colon; the mean from all fields was calculated and the results were expressed as a percentage of mild (grade I), moderate (grade II), and extensive (grade III) mucosal destruction of the entire colon. The crypt damage score was then calculated as previously described.16 Briefly, grades I, II, and III were given scores of one, two, and three respectively. In addition, the percentages of the respective injured surface areas for each field were scored on a scale of 1 = 10% up to 10 = 100%. The product of the two scores gave the crypt damage score for each field. Means for all fields were then calculated for each animal.
TGF-α RADIOIMMUNOASSAY
To determine the degree of TGF-α overexpression in the four groups, colonic tissue samples of transgenic mice and littermate controls (three per group) that did not receive DSS were pooled and frozen in liquid nitrogen immediately after death, cut into small pieces, and then homogenised on ice in 5 ml extraction buffer (1% trifluoroacetic acid (TFA), 5% formic acid, and 1% NaCl in 1 M HCl28) for one minute using a Tekmar homogeniser (Tekmar, Cincinnati, Ohio, USA). The mixture was then centrifuged at 3000 rpm for 10 minutes to remove debris, followed by high speed centrifugation at 55 000 rpm for 60 minutes. The lipid supernatant was removed, and the clear extract was loaded on to a Waters C18 Sep-Pak cartridge (Millipore Corp., Milton, Massachusetts, USA) which was equilibrated with 10 ml 100% acetonitrile followed by 10 ml 0.1% TFA. After loading, the cartridge was washed with 2 ml 10% acetonitrile in 0.1% TFA before elution of the TGF-α-containing fraction with 2 ml 40% acetonitrile in 0.1% TFA. Fractions were evaporated in a Speed-Vac concentrator (Savant Instruments Inc., Farmingdale, New York, USA) and redissolved in radioimmunoassay buffer. TGF-α was determined in aliquots of tissue extracts with a commercially available radioimmunoassay kit (Peninsula Laboratories, Belmont, California, USA). Protein concentrations of the clarified acidic extracts were determined as previously described.29 Results were expressed in pg/100 μg protein in the acidic extract. Recovery was determined by assaying colonic extracts of Balb/c mice to which a defined amount of human recombinant TGF-α (Fisher Scientific, Tustin, California, USA) had been added and was 82%. The detection limit of the assay was 8 pg/100 μg protein in the acidic extract.
ASSESSMENT OF PROLIFERATING CELLS USING ANTI-BROMODEOXYURIDINE (BrdU)
Mice of all four groups without colitis (three per group) received an intraperitoneal injection of 50 mg/kg BrdU (Sigma Chemical Co.) 120 minutes before being killed. Paraffin embedded sections at standard thickness were deparaffinised and digested with 0.1% protease solution (Sigma Chemical Co.), followed by treatment with 2 M HCl. Endogenous peroxidase was quenched with 3% H2O2. Slides were blocked with a 10% solution of normal horse serum in phosphate buffered saline, then incubated with anti-BrdU30 (Dako Corp., Carpentaria, California, USA), diluted 1:400 in 1% bovine serum albumin. After being washed, sections were incubated with biotinylated horse anti-mouse IgG and placed in peroxidase linked avidin-biotin complex (Vector Laboratories, Burlingame, California, USA), diluted 1:100 in 1% bovine serum albumin. Slides were then exposed to diaminobenzidine (Sigma Chemical Co.) substrate (10 mg diaminbenzidine, 20 ml phosphate buffered saline, 20 μ1 30% H2O2) for 10 minutes. Sections were counterstained with haematoxylin. The total number of proliferating cells per crypt was determined at a magnification of 1:100 in 20 longitudinal cuts of colonic crypts by two independent investigators blinded to the group. Results were expressed as the total number of BrdU positive cells per crypt.
MUCIN STAINING WITH ALCIAN BLUE
Tissue was fixed in zinc/formalin, embedded in paraffin, and cut into 7 μm thick sections. Slides were deparaffinised and hydrated to water. Acidic mucins were stained with alcian blue at pH 2.5, and sulphated mucins were stained with alcian blue at pH 1.0.31 The total number of stained goblet cells per crypt (acidic mucins) was determined at a magnification of 1:100 in 20 longitudinal cuts of colonic crypts by two independent investigators blinded to the group. Results were expressed as the total number of goblet cells with positive staining for alcian blue at pH 2.5 per crypt.
STATISTICAL ANALYSIS
Results are expressed as means (SE). Significance of differences was assessed by the Mann-Whitney U test. p<0.05 was considered to be statistically significant.
ETHICAL CONSIDERATIONS
The studies were approved by the animal care committee of the University of Wisconsin in accordance with the standards set down in the Animal Welfare Act and other federal, state, and local statutes and regulations relating to animals.
Results
TGF-α RADIOIMMUNOASSAY
Colonic extracts (pooled tissue of three animals per group) of TG+ and TG− mice contained 565 (in the acidic extract) and 67 pg TGF-α/100 μg protein respectively. The corresponding values for non-transgenic littermate controls were 64 and 76 pg/l00 μg protein in Non-TG+ and Non-TG− mice respectively.
ASSESSMENT OF DSS/WATER CONSUMPTION
Mean consumption of DSS/water per animal during the trial was 27.2 (1.7) ml in TG+ mice, 27.6 (1.6) ml in TG− mice, 22.5 (0.6) ml in Non-TG+ mice, and 29.0 (1.1) ml in Non-TG− mice. There was a statistically significant difference (p<0.05) between Non-TG+ animals and all other groups. Cumulative day to day analysis showed a difference in the last three days of the trial, when Non-TG+ mice drank significantly less water than mice of the other groups.
SEVERITY OF DSS INDUCED COLONIC INJURY
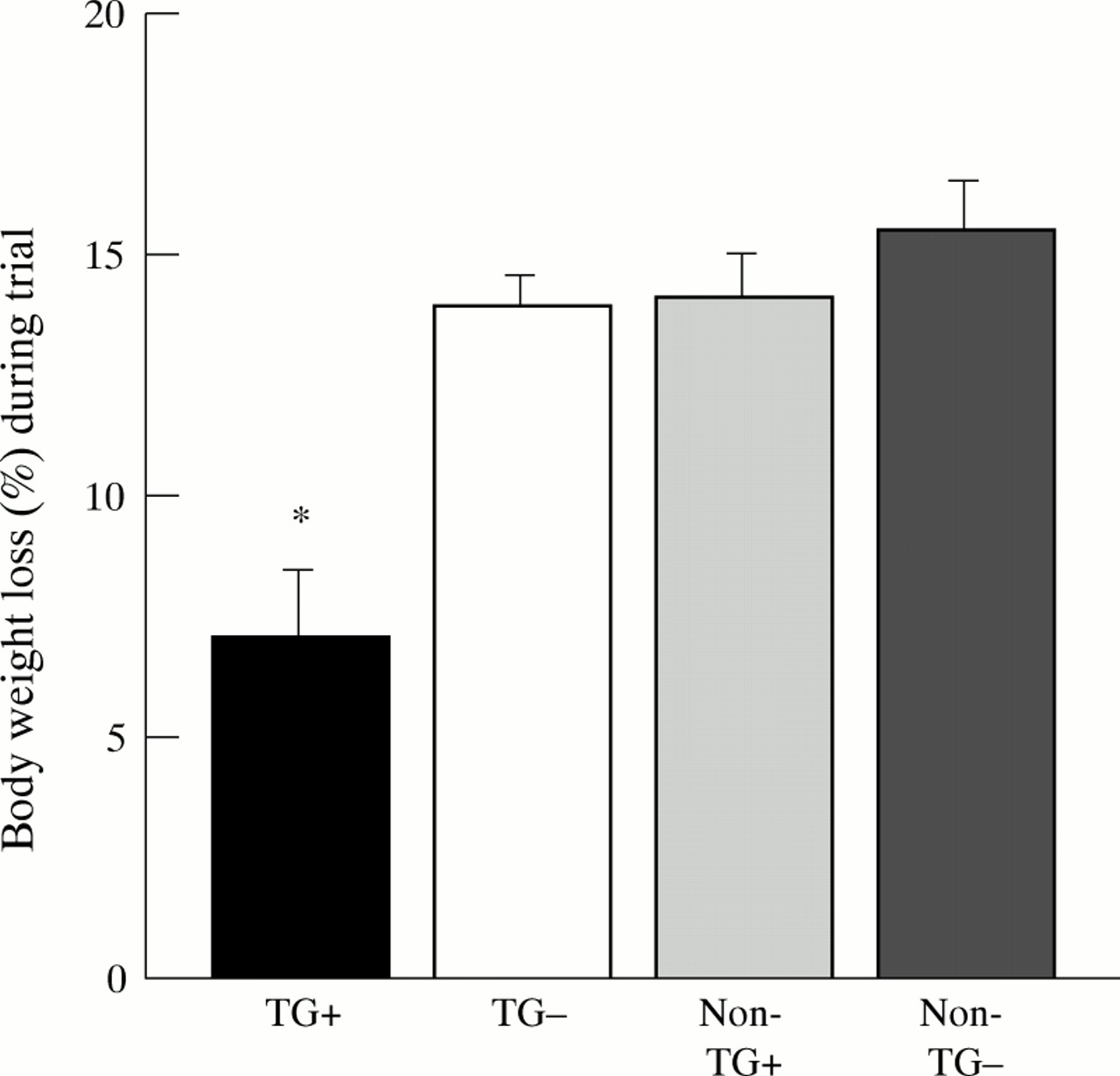
Body weight decreased in all groups with oral administration of 5% DSS in the drinking water (fig 1). Clinically, a progressive fall in body weight, profuse rectal bleeding, diarrhoea, and progressive lethargy was noted in the Non-TG−, Non-TG+, and TG− group after three or four days of DSS administration. Three animals of the TG− group died at the beginning of day six and were excluded from histological assessment (autolysis) and from statistical analysis. Loss of body weight and diarrhoea were also observed in TG+ mice towards the end of the trial but were much less pronounced. TG+ mice lost only 7.0% (2.2 g) of total body weight over the course of DSS treatment, whereas the corresponding values for the other groups were 13.8% (4.7 g) for TG− mice, 14.1% (5.1 g) for Non-TG+ mice, and 15.4% (5.6 g) for Non-TG− mice (fig 1). Considerable amounts of blood were seen in the colonic lumen of TG− mice, Non-TG+, and Non-TG− mice, and to a much lesser degree in the TG+ group (fig 2).
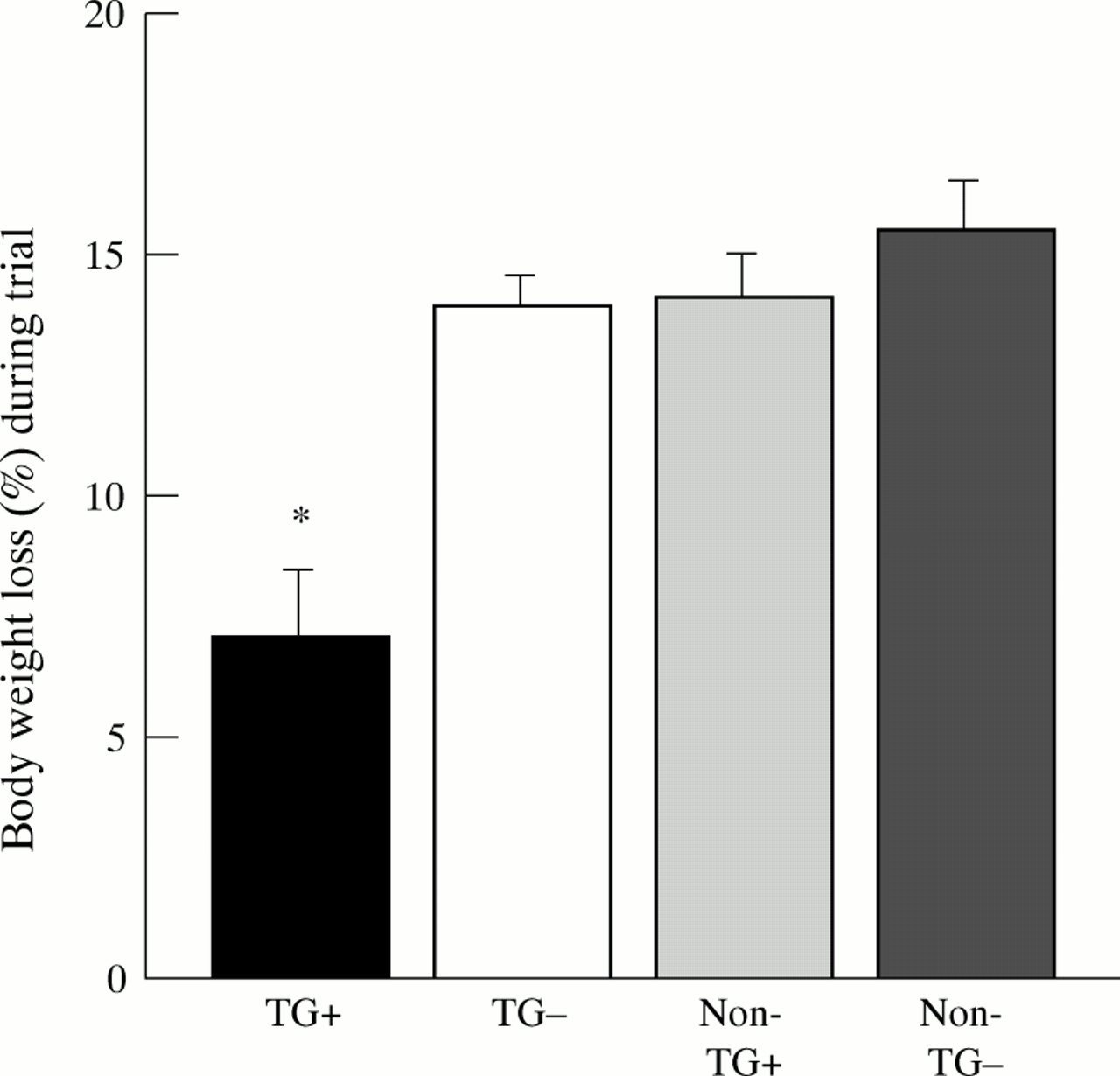
Decrease in body weight (expressed as a percentage of the initial weight) of transforming growth factor α (TGF-α) transgenic mice and littermate controls with and without zinc administration six days after induction of colitis. TGF-α transgenic mice without zinc (TG−), and littermate controls with (Non-TG+) and without zinc administration (Non-TG−) had a similar decrease in body weight, which was significantly more than that observed in zinc induced TGF-α transgenic mice (TG+) (n = 10 per group). *p<0.05 v other groups.

Blood content in transforming growth factor α (TGF-α) transgenic mice and littermate controls with and without zinc administration six days after induction of colitis. Colon blood content was graded from 0 to 3+ (0, no blood; 1+, blood in up to one third of the entire colon; 2+, blood in up to two thirds of the entire colon; 3+, blood in more than two thirds of the entire colon). There was significantly less blood in the colons of TGF-α transgenic mice with zinc induction (TG+) than in the three control groups (TG−, Non-TG+, Non-TG−). *p<0.05 v other groups.
Colonic length decreased significantly by 29.4 (2.2)% in Non-TG− mice, by 29.3 (2.1%) in Non-TG+ mice, by 29.3 (1.7)% in TG− mice, and by 14.6 (2.8)% in TG+ mice when compared with their respective control animals without colitis. The decrease was significantly less in TG+ mice (all p<0.05) compared with all other groups.
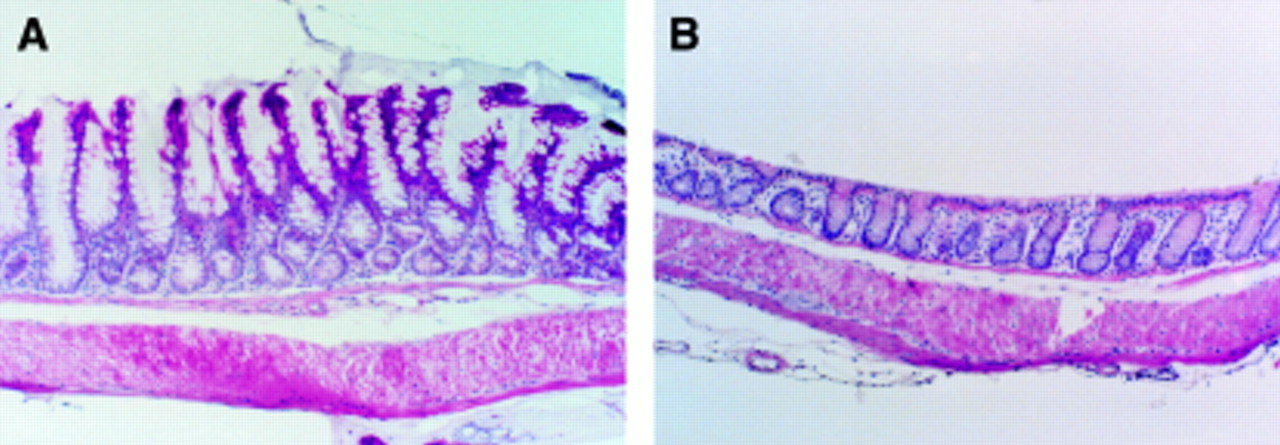
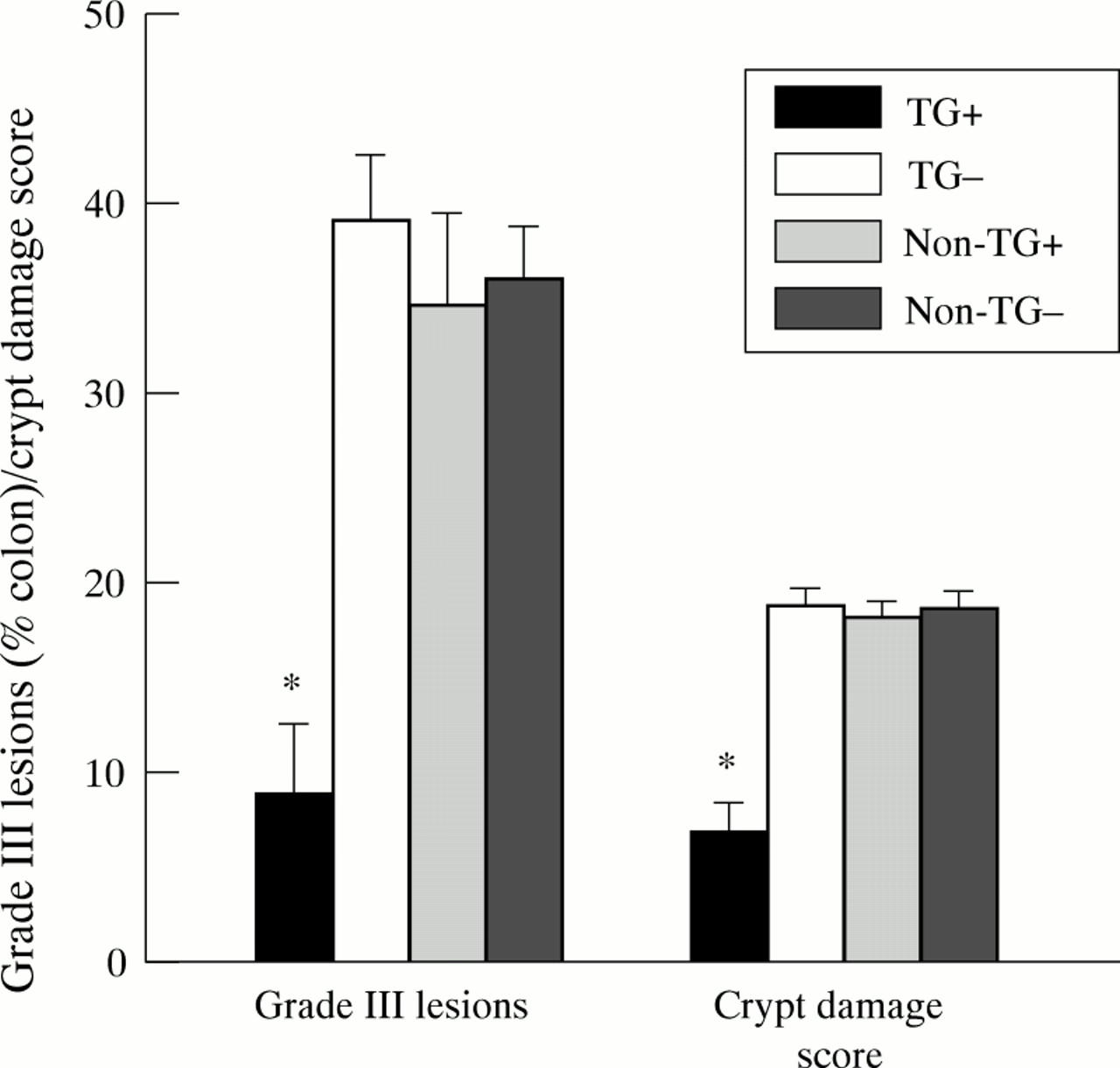
Microscopic evaluation of inflamed colons showed a more extensive and severe destruction of the mucosal glandular architecture in TG−, Non-TG+, and Non-TG− animals than TG+ mice. Marked hyperplasia was found in the transgenic mice induced by zinc with crypts forming villi (fig 3A). During colitis the destruction of the mucosal glands was characterised by increasing loss of the mucosal epithelial cells and crypts, initially at the bottom of the crypts and subsequently a complete loss of epithelial structures but not of the lamina propria. Mononuclear cells, including foamy macrophages, plasma cells, and lymphocytes, were observed in the affected areas. Neutrophils were also seen in the submucosa below the damaged mucosa. Evaluation of the colonic damage showed that TG−, Non-TG+, and Non-TG− mice had significantly more grade III (erosions/glandular destruction of the entire mucosa) lesions (39, 35 and 36% respectively) than TG+ mice (9%) (fig 4). Grade I lesions were 14.8 (1.2), 13.4 (0.1), 15.6 (1.9), and 13.5 (1.1)% in TG+, TG−, Non-TG+, and Non-TG− mice respectively. Statistically there were no significant differences in grade I lesions between the different groups. The corresponding values of grade II lesions were 13.5 (2.6), 28.9 (3.7), 31.4 (2.4), and 32.9 (2.1)% in TG+, TG−, Non-TG+, and Non-TG− mice respectively. Grade II lesions in TG+ mice were significantly different from those in all other groups (all p<0.05) but were not different among the control groups. The crypt damage score reflecting all lesions was 6.8 (1.5), 18.9 (0.8), 18.2 (1.0), and 18.7 (0.9), in TG+, TG−, Non-TG+, and Non-TG− animals respectively (fig 4). Crypt damage scores were significantly different between TGF-α transgenic mice receiving zinc (TG+) and all other control groups (all p<0.01) and were not different among the control groups (fig 4).
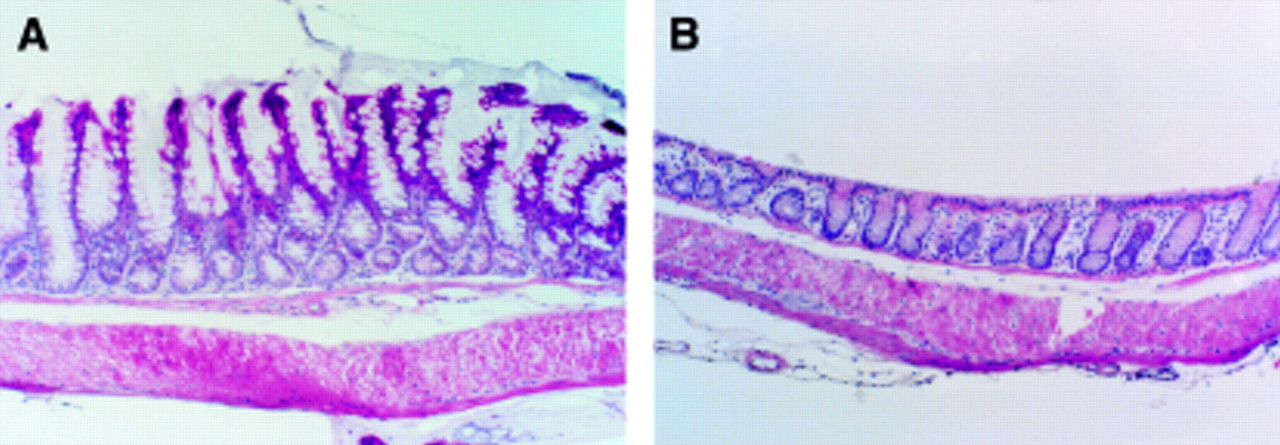
Histological findings in transforming growth factor α (TGF-α) transgenic mice (control groups without colitis) with (A) and without (B) zinc administration. Note the marked hyperplasia of the mucosa with increased cell number and increased crypt depth in TGF-α transgenic mice with zinc administration (A) compared with those without zinc administration (B). (Haematoxylin and eosin; original magnification × 40).

Crypt damage score and grade III lesions in dextran sodium sulphate induced colitis of transforming growth factor α (TGF-α) transgenic mice and littermate controls with and without zinc administration. Significantly more severe erosions/glandular destruction involving the entire mucosa (grade III) were seen in the three control groups (TG−, Non-TG+, and Non-TG−) than in TGF-α transgenic mice with zinc induction (TG+). *p<0.05 v other groups. Note that the crypt damage score reflecting all pathology of the glandular architecture (grade I–III) was also significantly different in control groups compared with zinc induced TGF-α transgenic mice.
Microscopic examination of the stomach and small intestine of all four colitis groups showed no evidence of mucosal injury or inflammation (data not shown). In control mice not given DSS, no weight loss, diarrhoea, or rectal bleeding was noted. No blood was detected in the opened colons and histologically no evidence of mucosal injury or inflammation was present. However, a marked hyperplasia of the colonic mucosa with massive mucin content was found in TG+ mice when compared with the other groups (fig 3A and B, fig 5A and B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mucin staining of colons (without colitis) in transforming growth factor α (TGF-α) transgenic mice with and without zinc administration. Acidic mucin staining was more pronounced (increased number of goblet cells and increased staining intensity) in TGF-α transgenic mice with zinc administration (A) than in those not given zinc (B). (Haematoxylin and eosin; original magnification × 80).
ASSESSMENT OF MUCOSAL PROLIFERATION (BrdU STAINING) IN MICE WITHOUT COLITIS
There was a greater than 100% increase in the number of BrdU labelled cells in the non-inflamed colonic tissue of TG+ mice (11.9 (0.6)) when compared with TG− (4.5 (0.4)), Non-TG+ (4.2 (0.3)), and Non-TG−(4.1 (0.5)) mice. This increase in BrdU staining was localised to the basal third of the crypts.
ASSESSMENT OF MUCIN STAINING
A marked increase in goblet cells that stained positively for acidic mucin with alcian blue at pH 2.5 was noted in TG+ mice (54.8 (0.7)) when compared with TG− (15.1 (0.4)), Non-TG+ (15.0 (0.4)), and Non-TG− (14.5 (0.4)) mice (fig 5) and was not different among the control groups. The increase in the number of goblet cells and the increase in staining were noted in TG+ mice with and without colitis. Goblet cells staining positive for acidic mucin extended along the entire length of the crypt throughout the entire colon. The number of goblet cells staining for sulphated mucin with alcian blue at pH 1.0 did not appear to be affected by the TGF-α overexpression.
Discussion
The results of this study show for the first time that genetically altered mice that overexpress TGF-α develop considerably less severe and extensive colonic mucosal injury in response to DSS administration than littermate controls without transgene expression. DSS, a sulphated polysaccharide, reproducibly induces acute and chronic colitis in mice,32 ,33 with features of ulcerative colitis, characterised by a T helper-2 response.34 ,35 This results in appreciable loss of body weight, rectal bleeding, a reduction in colonic length, and profound destruction of the colonic epithelium and glandular architecture. DSS is thought to induce mucosal injury and inflammation initially through a direct toxic effect on epithelial cells with subsequent activation of macrophages and T cells resulting in cytokine mediated cytotoxicity.34 ,36 We recently showed that mice lacking TGF-α (TGF-α knockout mice and wa-1 mice) have considerably increased susceptibility to DSS.16 We were also able to show that exogenous administration of TGF-α reduced the severity of DSS induced mucosal injury in wa-1 mice, an inbred strain with a TGF-α deficiency.16 However, it is not known whether in null mutation (knockout) strains compensatory processes generated during fetal or postnatal development are in place that may be partially responsible for the pathophysiological phenomena observed.18-20 In addition, since TGF-α is known to play an important role in the embryological development of mammals,21-26 adult knockout mice may lack not only TGF-α but also entire biological pathways that depend on TGF-α during development. The results of this study showing a pronounced decrease in DSS induced mucosal injury in TGF-α transgenic mice, in which transgene overexpression has been initiated in adults, underline and confirm our hypothesis that TGF-α is a key mediator of protection and/or repair mechanisms in the colonic mucosa.
The reduced susceptibility of mice overexpressing TGF-α to DSS colitis raises the possibility that the integrity of the colonic mucosal barrier is strengthened in these animals. It has been postulated that negatively charged sulphated polysaccharides such as DSS injure colonic epithelial cells and destroy intraepithelial tight junctions,37 together with haemodynamic alterations.38 Since colitis induced by sulphated polysaccharides, such as carrageenan, only develops in the presence of the normal bacterial flora of the colon,39 ,40 a permissive role for bacteria that may cross the mucosal barrier in the development of colitis is suggested. The unique bacterial milieu of the colon may also help to explain the previous observations that healing of dermal wounds41 and healing after tail amputation42 were not impaired in TGF-α knockouts when compared with their genetic controls whereas DSS induced colitis was enhanced. The diversity and abundance of colonic bacteria are dramatically different from those of skin, and bacterial invasion after DSS induced colonic mucosal injury may partially explain why the colon is so sensitive to TGF-α deficiency. It will be interesting to study, using TGF-α transgenic and TGF-α knockout mice, mucosal defence and repair in different parts of the gastrointestinal tract where the bacterial flora differs considerably.
We previously reported enhanced expression of TGF-α immunoreactivity after the induction of colitis.15 This supported the idea that TGF-α, a peptide that shares structural and functional homology with EGF,8-12 contributes to the preservation of colonic mucosal integrity in response to a variety of noxious agents. Studies in humans have shown that TGF-α, but not EGF, is present in colonic mucosal biopsy specimens from patients with inflammatory bowel disease.43 Interestingly, increased levels of TGF-α mRNA were present in samples of patients with inactive rather than active ulcerative colitis,43 raising the possibility that this peptide may be involved in epithelial cell hyperproliferation in the colon. The results of the BrdU staining in this study show the proliferative effect of TGF-α on colonic epithelial cells. Thus, in inactive colitis ulcerosa, activation of protection and/or repair mechanisms may be induced by increased expression of TGF-α. It would be interesting to evaluate the proliferation rate of the colonic mucosa together with the expression of TGF-α in inactive colitis ulcerosa compared with healthy individuals.
The presence of immunoreactivity for TGF-α within the Golgi apparatus as well as in goblet cell granules suggests a possible secretory function for TGF-α within the colon.17 Transgenic murine strains that overexpress TGF-α have been shown to contain increased gastric mucin.44 In the present study, the results of the mucin staining in animals with and without colitis show a markedly increased number of goblet cells and mucin content of the entire colonic mucosa. These findings together with the results of the BrdU staining suggest that TGF-α increases proliferation of goblet cells and increases mucin synthesis within goblet cells. These effects on overall mucin production by TGF-α may also contribute to the reduced extent and severity of DSS colitis in mice through mucus scavenging of toxic oxygen metabolites as described previously.45
Recently, it was reported that intestinal trefoil factors play a central role in the maintenance and repair of the intestinal mucosa.46 Mice lacking trefoil peptides showed considerably increased susceptibility to DSS induced injury,46 similar to that observed previously in TGF-α knockout and wa-I mice.16 In addition, diminished intestinal trefoil factor staining in TGF-α knockout mice has been described (A S Giraud, G A Cook, and N D Yeomans, personal communication, May 1996). Since trefoil peptides are co-expressed with mucin glycoproteins,47 ,48 it is possible that TGF-α stimulates intestinal trefoil factor synthesis and release from goblet cells, and that the preserving/healing properties of TGF-α are mediated through trefoil peptides.
One could argue that the observed morphological and functional alterations of the colonic mucosa in mice overexpressing TGF-α are just “pharmacological” effects and do not necessarily prove the importance of endogenous TGF-α in the maintenance of normal colonic mucosa. However, the results of the present study together with previous results on the high susceptibility of TGF-α deficient mice to DSS induced colitis16 strongly support our hypothesis that endogenous TGF-α is an important mediator of mucosal protection and/or repair mechanisms.
In conclusion, our studies show that transient induction of TGF-α in adult transgenic mice significantly reduces susceptibility to colitis induced by oral DSS. TGF-α is known to have a spectrum of biological activities. Its considerable proliferative effects on colonic mucosal cells, including goblet cells, as well as the enhanced staining for acidic mucins may explain the beneficial effects of TGF-α in providing mucosal healing and protection in the DSS colitis model. Our studies substantiate a key role for endogenous TGF-α as a mediator of mucosal protection and/or repair and suggest a potential therapeutic role for TGF-α in various inflammatory disorders such as inflammatory bowel disease.
Acknowledgments
This study was supported by the Inflammatory Bowel Disease Center, Harbor-UCLA Medical Center, Torrance, California, and the Crohn’s and Colitis Foundation of America (to V E E).