Article Text

Abstract
Objective The α4β7 integrin monoclonal antibody vedolizumab is hypothesised to be gut selective. Effects of vedolizumab on immune responses to parenterally or enterally administered antigens were investigated.
Design In this randomised, double-blind, placebo-controlled, phase I trial, healthy participants received a single intravenous dose of vedolizumab 750 mg (n=64) or placebo (n=63). After 4 days, participants began intramuscular hepatitis B vaccine (HBV; days 4, 32, 60) and oral cholera vaccine (OCV; days 4, 18) regimens. The study was designed to demonstrate a 15% non-inferiority margin for the between-group difference in the primary end point: percentage of participants with HBV seroconversion at day 74 (serum hepatitis B surface antigen (HBs) antibody titre ≥10 IU/L). OCV seroconversion at day 74 (>4-fold increase in serum cholera toxin (CT) antibodies) was a secondary end point.
Results A total of 56 (90.3%) placebo-treated and 54 (88.5%) vedolizumab-treated participants responded to HBV. Geometric mean anti-HBs titres were similar for placebo (114.4 IU/L) and vedolizumab (129.6 IU/L) at day 74. A total of 60 (96.8%) placebo-treated and 52 (82.5%) vedolizumab-treated participants responded to OCV at day 74. Geometric mean anti-CT IgG levels were higher for placebo than for vedolizumab at day 74 (9210.08 vs 3007.8 ELISA Units (EU)/mL) and day 32 (11629.3 vs 1575.4 EU/mL). Anti-CT IgA results were similar. Adverse events were consistent with previous experience. One serious adverse event (spontaneous abortion) was reported for placebo.
Conclusions Vedolizumab did not alter the response to parenterally administered antigens but reduced the response to oral antigens, demonstrating its gut-selective mechanism of action.
Trial registration number NCT Number: 01981616; EudraCT Number: 2011-001874-24.
- Gut Immunology
- Antibody Targeted Therapy
- Inflammatory Bowel Disease
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
-
Vedolizumab, a humanised α4β7 integrin monoclonal antibody in development for the treatment of ulcerative colitis (UC) and Crohn's disease (CD), downregulates gut inflammation by inhibiting intestinal T-lymphocyte trafficking.
-
Systemic inflammatory bowel disease therapies (corticosteroids, thiopurines, tumour necrosis factor α (TNF-α) antagonists) are associated with increased risk of serious infection. The systemically active anti-α4 integrin therapy natalizumab carries a risk of progressive multifocal leukoencephalopathy, a serious central nervous system viral infection thought to arise from reduced T-cell trafficking to the brain.
-
Placebo-controlled phase III studies have demonstrated that the overall rates of serious infections were similar in vedolizumab-treated and placebo-treated patients with UC or CD.
-
Impaired responses to hepatitis B vaccine have been observed in patients treated with TNF-α antagonists.
What are the new findings?
-
Antibody titres to parenterally administered hepatitis B vaccine did not appreciably differ over time between participants treated with placebo and those treated with vedolizumab.
-
The overall magnitude of the humoral response to enterically administered oral cholera vaccine was significantly reduced in vedolizumab-treated participants, but not in those who received placebo.
-
Vedolizumab did not alter the response to parenterally administered antigens, but did reduce the response to oral antigens, supporting the hypothesis that its mechanism of action is selective for the gastrointestinal system.
How might it impact on clinical practice in the foreseeable future?
-
These data support the premise that selective immunomodulatory agents such as vedolizumab hold great promise for the treatment of inflammatory bowel diseases.
-
Our results are of considerable importance for patients who may be undergoing treatment with vedolizumab and are in need of vaccination, particularly with oral or mucosal vaccines.
Introduction
The α4β7 integrin, expressed on leucocyte subpopulations, represents a key determinant of gut mucosal immunity.1 The principal adhesion ligand of α4β7, mucosal addressin cell adhesion molecule-1 (MAdCAM-1), is preferentially expressed on high endothelial venules at sites of lymphocyte extravasation in gastrointestinal mucosa and associated lymphoid tissue. Binding of MAdCAM-1 by lymphocytes expressing activated α4β7 mediates migration of these cells into tissues.2 ,3 Immunological correlates of antagonising this interaction in humans have not been described previously. Vedolizumab, a humanised monoclonal IgG1 antagonist of the α4β7 integrin currently in development for the treatment of ulcerative colitis (UC) and Crohn's disease (CD),1 selectively downregulates gut inflammation by inhibiting intestinal T-lymphocyte trafficking.
Conventional treatments for UC and CD, such as corticosteroids, thiopurines (eg, azathioprine, 6-mercaptopurine) and tumour necrosis factor α (TNF-α) antagonists, act systemically and are associated with an increased risk of serious infection.4–6 Likewise, the safety of the systemically active anti-α4 integrin therapy natalizumab is compromised by the risk of progressive multifocal leukoencephalopathy, a serious central nervous system viral infection that is thought to originate from interference with T-cell trafficking to the brain.7 In contrast, analysis of safety data from placebo-controlled phase III studies of vedolizumab demonstrated that the overall rates of serious infections were similar in vedolizumab-treated and placebo-treated patients. Results from these controlled human studies have suggested benefits of vedolizumab in treating UC and CD.8–11
The mechanism of action of vedolizumab is hypothesised to be restricted to the gastrointestinal tract, not affecting immune responses outside of the gastrointestinal tract, which might be expected to positively affect its safety profile. To test this hypothesis, the current immune challenge study was conducted in healthy participants to assess the immune responses to two specific vaccine series, a hepatitis B vaccine (HBV) (a parenterally administered antigen) and an oral cholera vaccine (OCV) (an enterally administered T-cell-dependent antigen),12 after administration of a single intravenous dose of vedolizumab or placebo. We hypothesised that humoral responses to HBV would be similar between vedolizumab- and placebo-treated participants, whereas responses to OCV would be attenuated with vedolizumab. This gut-selective immune modulation is an intriguing premise that, if achievable, holds great potential as a strategy for the treatment of UC and CD.
Materials and methods
Study design
The final protocol, amendments and informed consent forms were reviewed and approved by the independent ethics committee at the study site. The study was conducted in compliance with the protocol, Good Clinical Practice guidelines, and applicable regulatory requirements (including the International Conference on Harmonisation guidelines), and in accordance with the ethical principles in the Declaration of Helsinki. Written informed consent was obtained from all participants at the time of enrolment.
In this phase I, randomised, double-blind, placebo-controlled, parallel-group, single-centre (ICON Development Solutions, Manchester, UK) non-inferiority study, 127 healthy participants were randomly assigned in a 1:1 ratio to receive a single intravenous dose of vedolizumab 750 mg or placebo. This dose of vedolizumab was chosen to ensure adequate serum drug concentrations and complete α4β7 receptor saturation in peripheral blood throughout the study period, including key time points (ie, up to day 74). Pharmacodynamic and pharmacokinetic measurements were taken to ensure that adequate levels were achieved.
The study included a screening period (days −28 to −1), followed by a treatment period (days 1–60), during which all participants received a single dose of either vedolizumab 750 mg or placebo (day 1), an intramuscular dose of HBV on days 4, 32 and 60, and an oral dose of OCV on days 4 and 18. An observation period (days 61–127) followed treatment (figure 1).

Study design. ⋆, Enrolment and dosing on day 1; a participant was considered to be enrolled in the study when he/she had received any amount of study drug. ▴, Hepatitis B vaccine administered on days 4, 32 and 60. ♦, Cholera vaccine administered on days 4 and 18. ET, early termination.
Vaccine responses were assessed until day 74. Safety was assessed up to day 127 by monitoring adverse events, vital signs and laboratory variables. Primary and key secondary end points were HBV and OCV seroconversion rates, respectively. For HBV, seroconversion was defined as a serum hepatitis B surface antigen (HBs) antibody concentration of ≥10 IU/L. For OCV, seroconversion was defined as a greater than fourfold increase from baseline in the cholera toxin (CT) antibody titre (IgG, IgM or IgA).13 ,14 Serum HBs and CT antibody levels were determined on day 1 (predose) and on days 18, 32, 60 and 74. The primary assessment was conducted on day 74. The total study duration, including screening, was 5 months.
Participants
Eligible participants were healthy men or women between the ages of 18 and 39 years with a body mass index between 18 and 32 kg/m2, inclusive. Individuals were excluded if they had: known exposure to hepatitis B virus; known prior hepatitis B vaccination irrespective of the number of doses received or previous employment in a healthcare setting; seropositivity for prior hepatitis B infection or hepatitis B vaccination during the screening period; or known exposure to cholera or prior OCV exposure irrespective of the number of doses received.
Randomisation and masking
Participants were allocated to treatment using blocked randomisation (block size of 2). Randomisation was centralised using an interactive voice response system. Investigational pharmacy staff members were the only site personnel who were aware of the treatment assignment. All other study site personnel and the study participants were blinded.
Vaccines
The HBV (HBvaxPRO; Sanofi Pasteur MSD, Maidenhead, Berkshire, UK) is an intramuscular recombinant component vaccine approved for use in the European Union and elsewhere.15 The OCV (Dukoral; Crucell Vaccines, Toronto, Ontario, Canada (a division of Janssen)) is a combination of killed bacterial cells and the recombinant B subunit of the CT that is approved in some European countries and Canada for the prevention of traveller's diarrhoea.16
Assessment of immune response
Blood samples were collected for assessment of the immune response in serum to the vaccines at screening (HBs antibodies) on day 1 (predose; CT antibodies) and on days 18, 32, 60 and 74 (HBs and CT antibodies). Faecal and saliva samples were obtained for assessment of secretory IgA on day 1 (predose) and on days 18, 32 and 60; an additional saliva sample for assessment of secretory IgA was collected on day 74.
Pharmacokinetic/pharmacodynamic assessments
Blood samples for the determination of serum vedolizumab concentrations were collected on day 1 (pre- and post-dose) and on days 18, 32, 60, 74 and 127. Serum vedolizumab concentrations were measured using an ELISA. Blood samples for the determination of MAdCAM-1-Fc binding (indicative of the extent of cellular α4β7 integrin binding by vedolizumab) were collected on day 1 (predose) and on days 4, 18, 32, 60, 74 and 127. This binding was analysed using soluble MAdCAM-1-Fc and detected by flow cytometry using a fluorescently labelled anti-mouse Fc.
Statistical analysis
Sample size was calculated to demonstrate a non-inferiority margin of 15% with the lower bound of the one-sided 95% CI for the difference in the proportions of participants with HBV seroconversion between the vedolizumab and placebo groups, assuming a 90% seroconversion rate. Assuming a 90% evaluability rate, up to 125 participants were needed for randomisation (table 1).
Power estimates for primary and key secondary efficacy analyses
For the primary objective, to determine the effect of vedolizumab on the immune response to HBV at day 74, a point estimate and 95% CI for the between-group difference in the proportions of participants having an immune response were obtained for the per-protocol population. The lower bound of the 95% CI was compared with the prespecified non-inferiority margin of −15%. The same analysis was performed for the key secondary objective, to determine the effect of vedolizumab on the immune response to OCV at day 74 (table 1).
Results
Participant disposition and baseline characteristics
The trial was conducted from 13 September 2011 to 13 July 2012. Of 332 healthy individuals screened for eligibility, 127 were randomly assigned to receive a single intravenous dose of either placebo (n=63) or vedolizumab 750 mg (n=64) (figure 2). A total of 125 participants completed the study; one participant in the placebo group was withdrawn from the study because of an adverse event (injury to skin on the elbow), and one participant in the vedolizumab group was withdrawn because of protocol deviations (participant was unable to attend further study visits after study drug dosing). Four participants were excluded from the per-protocol analysis because of protocol deviations. Reasons for exclusion were non-receipt of either vaccine as scheduled (n=1 each for placebo and vedolizumab) and non-measurable serum vedolizumab concentration at day 74 (n=2 for vedolizumab). Treatment groups were generally similar with regard to demographic and other baseline characteristics (table 2).
Demographic and other baseline characteristics: intention-to-treat population
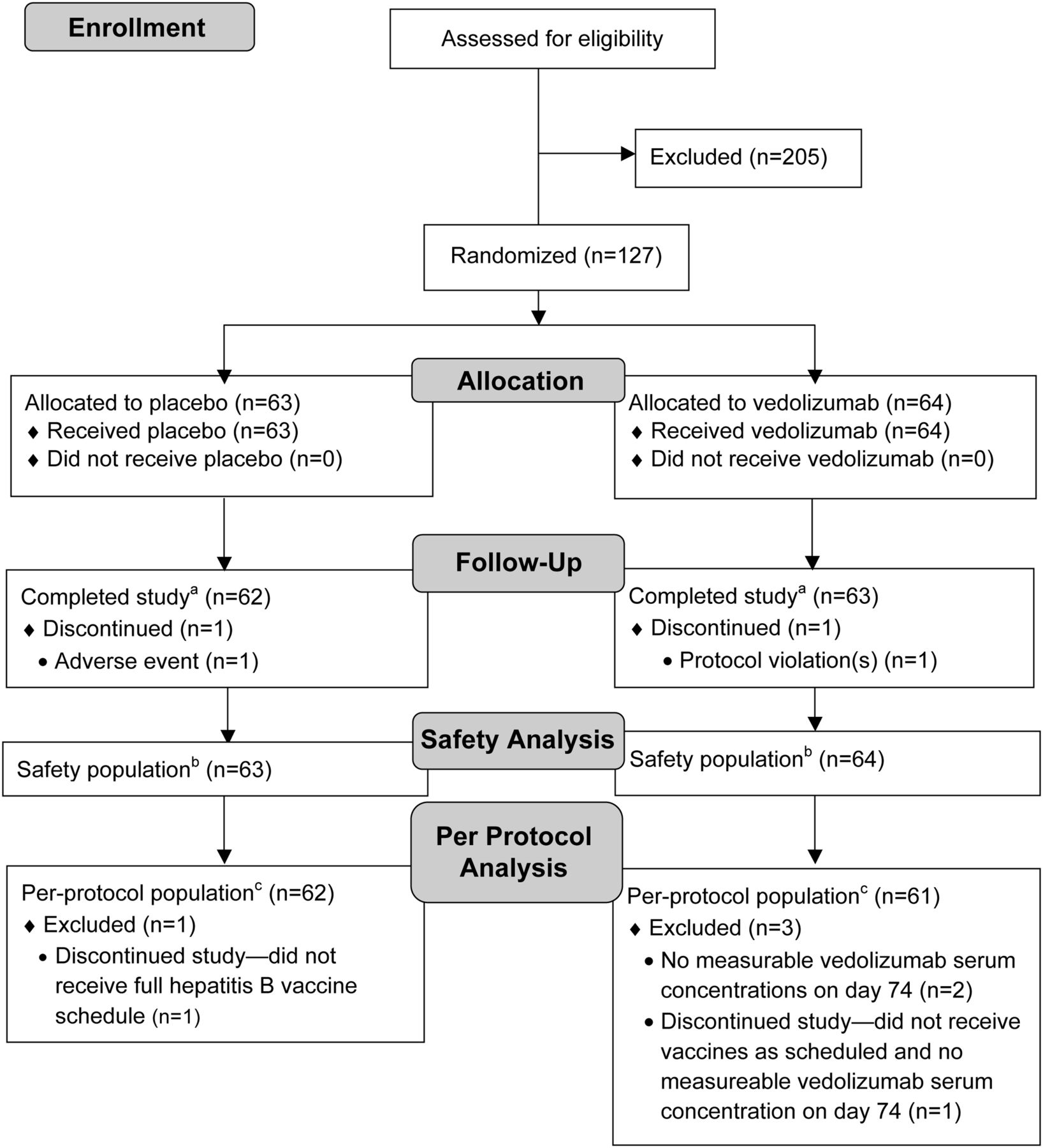
Participant flow diagram. aCompleted all day 127 assessments. bAll participants who received any amount of blinded study drug based on what they actually received. cAll participants without any major protocol deviations.
Serum vedolizumab concentrations and α4β7 saturation
The mean serum vedolizumab concentration in vedolizumab-treated participants was 20.1 µg/mL at day 74 (figure 3). This concentration of vedolizumab was associated with maximal saturation of α4β7 on peripheral blood CD4+CD45RO+ T cells (figure 3).

Inhibition of mucosal addressin cell adhesion molecule-1 (MAdCAM-1)-Fc binding to peripheral blood CD4+CD45RO+ T cells. Peripheral blood samples were taken at baseline (day 1 predose) and on days 4, 18, 32, 60, 74 and 127 and examined for ability to bind soluble MAdCAM-Fc. The n values represent the total number of healthy participants on day 1. Sample sizes on subsequent study days were as follows: day 4, n=53 (placebo) and n=55 (vedolizumab); day 18, n=54 (placebo) and n=55 (vedolizumab); day 32, n=61 (placebo) and n=62 (vedolizumab); day 60, n=61 (placebo) and n=61 (vedolizumab); day 74, n=61 (placebo) and n=62 (vedolizumab); day 127, n=60 (placebo) and n=62 (vedolizumab). Mean serum vedolizumab (VDZ) concentrations are shown in the table below the graph.
Immune responses to HBV and OCV
Of the 123 participants who received study drug and were included in the per-protocol population analysis, 56 of 62 (90.3%) in the placebo group and 54 of 61 (88.5%) in the vedolizumab group showed HBV seroconversion at day 74 (absolute difference, −1.8%; 95% CI −12.7% to 9.1%). Because the lower bound of the 95% CI is contained within the prespecified margin of −15%, the non-inferiority criterion was met for this end point. The time course of serum IgG response to HBV, as gauged by HBs antibody titres, is shown in figure 4A. The HBs antibody titres did not appreciably differ over time between the placebo group (median area under the concentration–time curve (AUC)day 1–74=1598 IU/L/day) and the vedolizumab group (median AUCday 1–74=2116 IU/L/day) (p=0.99; Wilcoxon two-sample test). At day 74, 2 weeks after the final immunisation, geometric mean (% coefficient of variation) HBs antibody titres were 114.4 (539.6) IU/L and 129.6 (359.3) IU/L for the placebo and vedolizumab groups, respectively.
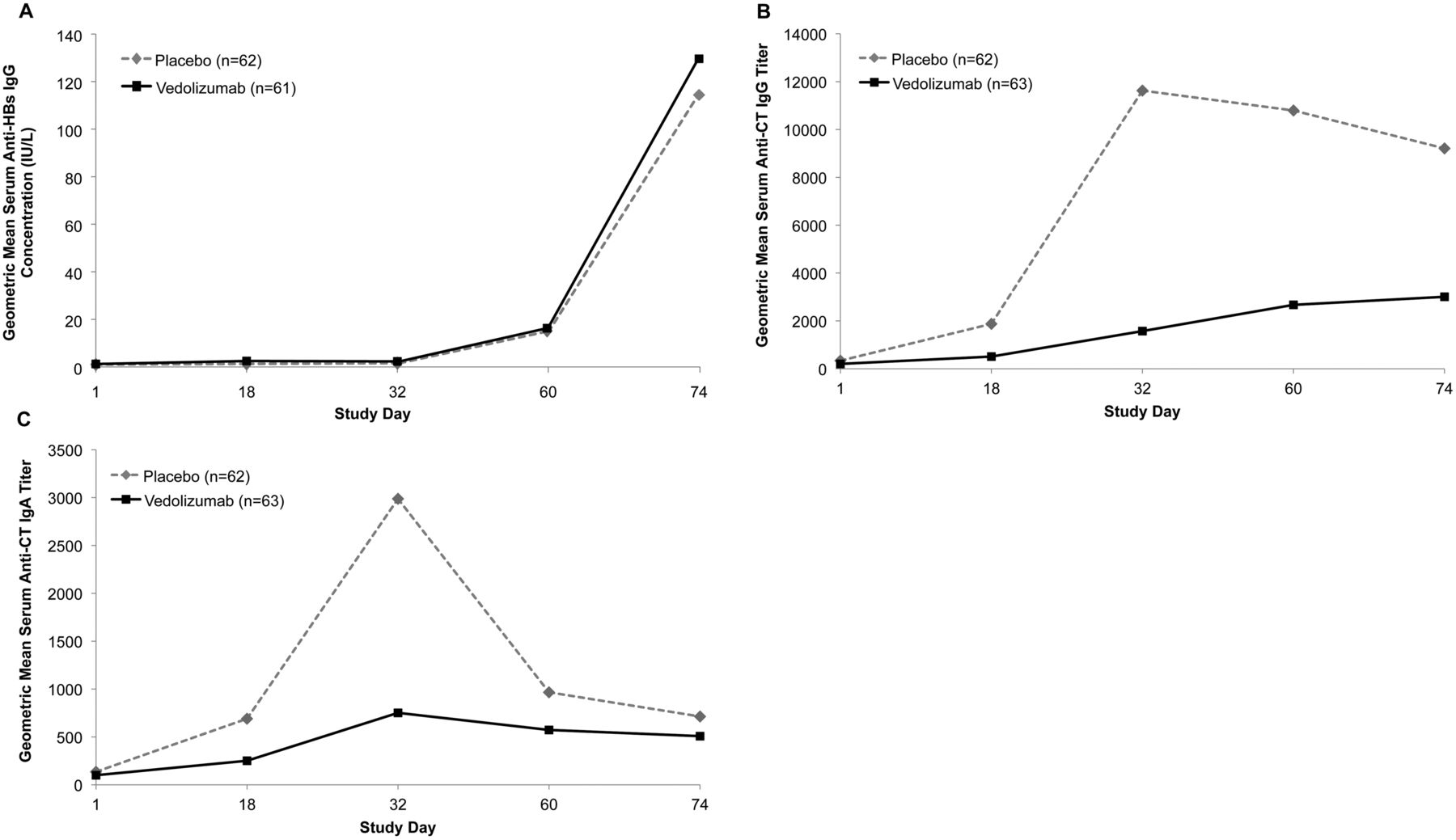
Kinetic evaluation of serum antibody responses to vaccine antigens. Blood samples were collected at baseline (day 1 predose) and on days 18, 32, 60 and 74, and serum (A) anti-(hepatitis B surface antigen (HBs)) IgG titres, (B) anti-(cholera toxin (CT)) IgG titres and (C) anti-CT IgA titres were measured. The geometric mean (% coefficient of variation) anti-HBs IgG titres on day 74 were 114.4 (539.6) IU/L for placebo and 129.6 (359.3) IU/L for vedolizumab. The geometric mean (% coefficient of variation) anti-CT IgG titres on day 32 were 11629.4 (420.9) EU/mL for placebo and 1575.4 (464) EU/mL for vedolizumab. The geometric mean (% coefficient of variation) anti-CT IgA titres on day 32 were 2987.1 (258.8) EU/mL for placebo and 751.8 (230.3) EU/mL for vedolizumab.
In contrast with HBV (for which the accepted standard for protective immunity is an initial serum HBs antibody titre of >10 IU/L),17 OCV is not associated with an accepted clinical standard of protective immunity; however, an increase from baseline of more than fourfold in IgM, IgA or IgG levels is considered a positive vaccine response.13 ,14 At day 74, OCV response rates for all evaluable participants were 96.8% and 82.5% in the placebo and vedolizumab groups, respectively (absolute difference −14.2%; 95% CI −24.6% to −3.9%). The entire 95% CI for the difference excludes 0, whereas the lower bound falls outside the prespecified margin of −15%; thus, OCV response in vedolizumab-treated participants did not meet the non-inferiority criterion. Time courses of serum IgG and IgA responses to OCV are shown in figures 4B, C respectively.
Although the percentage of OCV responders was not markedly different between treatment groups, the overall magnitude of the humoral response was significantly reduced in vedolizumab-treated participants. The AUCday 1–74 analyses showed significant differences between the vedolizumab and placebo groups for both serum IgG (median AUCday 1–74=124 384 and 736 610 titre·day, respectively) and serum IgA (median AUCday 1–74=42 952 and 118 868 titre·day, respectively) (both p<0.0001; Wilcoxon two-sample test).
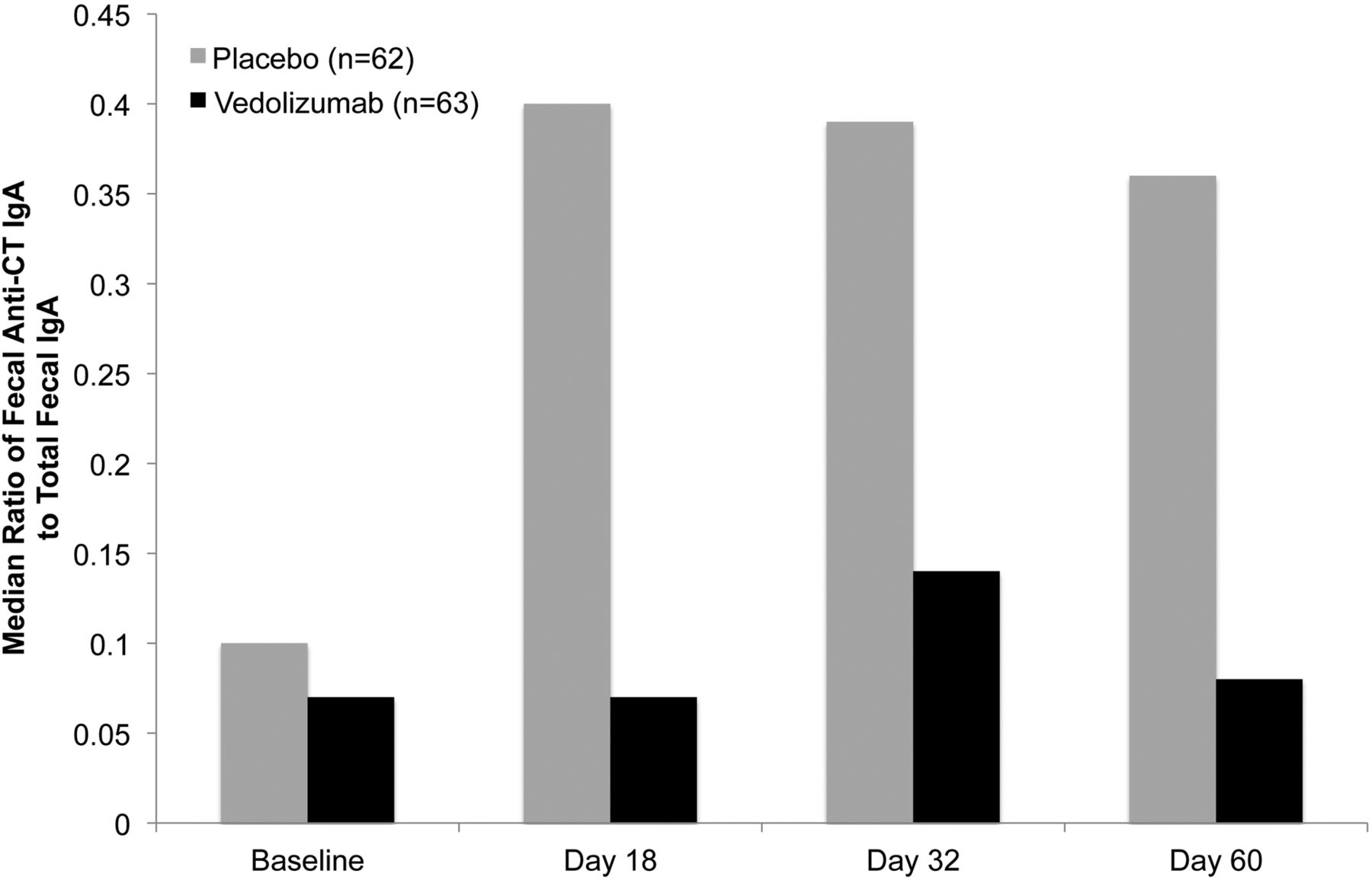
The secretory IgA response to CT was investigated in saliva and faeces. In saliva, no difference in anti-CT IgA, as a percentage of total IgA, was observed between the placebo and vedolizumab groups. However, the median ratio of faecal anti-CT IgA to total IgA in stool was severalfold higher with placebo than with vedolizumab (figure 5), and, considering the data available at days 1, 18, 32 and 60, this ratio peaked at day 18 for placebo and day 32 for vedolizumab.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Median ratio of anti-(cholera toxin (CT)) IgA to total IgA in faeces. Faeces were collected at baseline (day 1 predose) and on days 18, 32 and 60, and anti-CT secretory IgA titres were measured by ELISA in both the vedolizumab and placebo groups. The median ratio (25th, 75th centile) on day 32 was 0.39 (0.17, 0.99) for placebo and 0.14 (0.03, 0.2) for vedolizumab. The n values represent the total number of healthy participants on day 1. Non-adherence to faecal sample collection procedures for some participants led to varied n values at subsequent time points: day 18, n=21 (placebo) and n=26 (vedolizumab); day 32, n=40 (placebo) and n=23 (vedolizumab); day 60, n=32 (placebo) and n=22 (vedolizumab). Results are the median ratios of anti-CT IgA to total IgA in faeces multiplied by 100.
Safety
At least one treatment-emergent adverse event was experienced by 41 of 64 participants (64%) in the vedolizumab group and 50 of 63 participants (79%) in the placebo group (table 3). Infections were experienced by 20 of 64 (31%) and 24 of 63 (38%) participants in the vedolizumab and placebo groups, respectively. One serious adverse event (spontaneous abortion) was experienced by a placebo-treated participant.
Most commonly reported (≥3 participants) treatment-emergent adverse events: safety population
Discussion
This is the first study in humans to examine the effects of a lymphocyte trafficking antibody on local and systemic immunological priming, demonstrating a gut-selective immune effect. Antibody responses to HBV and OCV were measured in participants receiving the α4β7 integrin monoclonal antibody vedolizumab or placebo. In vedolizumab-treated participants, responses to HBV remained intact, whereas those to OCV were reduced, suggesting that the immunomodulatory effects of targeting the α4β7 integrin are selective to the gastrointestinal tract. Collectively, these data build on current understanding of the gut selectivity of the α4β7 pathway and support the hypothesis that the immunomodulatory effects of vedolizumab, a specific antagonist of α4β7 integrin, are restricted to the intestine.
The T-cell-dependent vaccines evaluated here were chosen to identify differences in humoral immune responses between parenteral and orally administered antigens. The HBvaxPRO vaccine, which contains the recombinant hepatitis B virus surface antigen, is given intramuscularly.15 Therefore, the humoral response generated by this vaccine is independent of mucosal immunological priming. In contrast, the Dukoral vaccine is a mixture of inactivated Vibrio cholerae bacteria and the recombinant B subunit of CT.16 ,18 The humoral immune response generated by this oral vaccine is initiated in the intestinal mucosa,18 and therefore was expected to be sensitive to blockade of lymphocyte trafficking to this compartment by vedolizumab.
Accordingly, whereas we observed no effect of vedolizumab on the percentage of participants with HBV seroconversion at day 74 or on the geometric mean HBs antibody titres, the response to OCV was substantially reduced in the vedolizumab group without being entirely abrogated. Although the difference in OCV response rates between the vedolizumab and placebo groups was modest but statistically different (−14.2%), it is notable that this difference for OCV did not meet the non-inferiority criterion of the prespecified comparison for the primary end point of hepatitis B seroconversion, which the study was powered to detect. The effect of vedolizumab on OCV response, though, was more evident when the magnitude of antibody response to OCV was assessed longitudinally. Whereas the rate of response to OCV in the placebo group was generally consistent with those from previously reported studies,13 ,19 ,20 serum IgG and IgA titres and the secretory IgA response to immunisation with OCV were lower after vedolizumab administration. These observations support the presence of a gut-selective effect of vedolizumab on T-cell-dependent vaccine responses. Whether vedolizumab exerts effects on other T-cell responses, including the induction of tolerance, is currently unknown and warrants further investigation.
The serum IgA results in the current study are consistent with recently published β7 integrin knockout mouse data.21 According to Schippers et al,21 the β7 integrin knockout mouse was unable to mount an IgA response after mucosal challenge. Of interest, though, while the clinical effect of blocking mucosal immunisation included a decrease in anti-CT IgG, this was not observed in the β7 integrin knockout, suggesting differences between mouse and human mucosal immune priming caused by either intrinsic or extrinsic factors.
Vaccine efficacy in individuals receiving anti-TNF-α therapies for immune-mediated disorders has been examined previously.22 Indeed, impaired response to HBV has been described in patients with rheumatoid arthritis receiving etanercept monotherapy.23 Other studies have yielded similar results.24 ,25 However, vaccination is generally considered worthwhile, as protective immunity can be achieved in most patients receiving these therapies.22 ,26
Although this study used a double-blind, placebo-controlled design, there are limitations that should be mentioned. The study participants were healthy volunteers, which may not adequately reflect the population (patients with UC or CD) for whom vedolizumab is intended. However, as the purpose of the study was to test the gut-homing hypothesis and the impact of inhibition of the α4β7–MAdCAM-1 interaction on immune priming, a healthy population was used to decrease the overall variability in vaccine responses due to variability in disease state. In addition, a single 750 mg dose of vedolizumab was administered in this study, in contrast with the multiple-dose regimen proposed for patients with UC or CD. It was anticipated that this 750 mg dose would ensure therapeutic drug concentrations at the day 72 primary end point, thus maintaining clinical exposure during the immune response to the vaccines. Indeed, the concentration of vedolizumab at day 72 was approximately twice as high as the 10 μg/mL serum trough concentration that has been shown to be efficacious in the phase III GEMINI trials.10 At day 127, serum vedolizumab concentrations were still sufficient to saturate α4β7 on peripheral T cells. Thus, sufficient vedolizumab was present at day 74 to adequately represent clinically efficacious levels of vedolizumab, ensuring that the results were not a consequence of inadequate drug concentrations at the primary end point or throughout the response period. The functionally long-lasting α4β7 blockage observed is probably due to the fact that low amounts of antibodies are required to saturate receptors. This long-lasting effect is not regarded as a safety concern since higher levels of antibody would be required to achieve therapeutic efficacy than that required to saturate the receptors. On the basis of the demonstrated clinical safety of vedolizumab, it is highly unlikely that the long-lasting α4β7 saturation would result in protracted impairment of gut immunity.
This is the first study to investigate potentially contrasting effects of a gut-selective therapeutic monoclonal antibody on the induction of mucosal and non-mucosal immunity. These results may raise questions about whether local immunosuppressive effects of targeting the α4β7 integrin could, in theory, impair host defences to oral antigens and predispose patients to enteric infections. To date, however, clinical studies have shown no evidence of increased risk of enteric infections with use of vedolizumab.8 ,10 ,11 ,27 These clinical findings may reflect the fact that the gut immune response, as demonstrated in this study, is attenuated—rather than entirely abrogated—by vedolizumab. We speculate that this level of immune modulation may be adequate to exert therapeutic effects without compromising host immune defences.
Acknowledgments
We wish to thank Irving Fox, MD, of Takeda Pharmaceuticals International Co and Robert J Gerety, MD, PhD, contract scientific consultant, for assistance with the study design, and Ralf Clemens, MD, PhD, of Takeda Pharmaceuticals International Inc, for critical review of the manuscript. Editing support was provided by Elizabeth Barton, MS, and Stefanie Dorlas, BMath, of MedLogix Communications, LLC, Manel Valdes-Cruz of Takeda Pharmaceuticals International Inc, and Elisabeth R Wann, PhD, of Wann Medical Communications, LLC.
References
Footnotes
-
Contributors TW, TL, SS, JP, MFP, BGF and AP were involved in the study design and/or collection of data. SS and YW were involved in the statistical analysis of data. All authors contributed to the writing and reviewing of all drafts of the manuscript. All authors approved the final, submitted version of the manuscript.
-
Funding This study was funded by Millennium Pharmaceuticals. Editing support was funded by Takeda Pharmaceuticals International Inc.
-
Competing interests TW is an employee of Takeda Pharmaceuticals International Co with equity stake. TL is a contractor who has received income from Takeda Pharmaceuticals International Co. SS is an employee of Takeda Pharmaceuticals International Co with equity stake. YW is a contractor who has received income from Takeda Pharmaceuticals International Co. JP is a former employee of Takeda Pharmaceuticals International Co. BGF is a consultant and advisory board member for Takeda. AP is an employee of Takeda Pharmaceuticals International Inc, with equity stake. MFP has no conflicts of interest to report.
-
Ethics approval Independent ethics committee at the study site.
-
Participant consent Obtained.
-
Provenance and peer review Not commissioned; externally peer reviewed.