Article Text

Abstract
Background Hepatic stellate cells play a key role in the pathogenesis of hepatic fibrosis.
Aims To examine the inhibitory effect of oestradiol on stellate cell activation.
Methods In vivo, hepatic fibrosis was induced in rats by dimethylnitrosamine or pig serum. In vitro, rat stellate cells were activated by contact with plastic dishes resulting in their transformation into myofibroblast-like cells.
Results In the dimethylnitrosamine and pig serum models, treatment with oestradiol at gestation related doses resulted in a dose dependent suppression of hepatic fibrosis with restored content of hepatic retinyl palmitate, reduced collagen content, lower areas of stellate cells which express α smooth muscle actin (α-SMA) and desmin, and lower procollagen type I and III mRNA levels in the liver. In cultured stellate cells, oestradiol inhibited type I collagen production, α-SMA expression, and cell proliferation. These findings suggest that oestradiol is a potent inhibitor of stellate cell transformation.
Conclusion The antifibrogenic role of oestradiol in the liver may contribute to the sex associated differences in the progression from hepatic fibrosis to cirrhosis.
- hepatic stellate cells
- hepatic fibrosis
- oestradiol
- α smooth muscle actin
- retinyl palmitate
Abbreviations
- SMA
- smooth muscle actin
- ECM
- extracellular matrix
- DMN
- dimethylnitrosamine
- PS
- pig serum
- RP
- reinyl palmitate
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- SDS
- sodium dodecyl sulphate
- DMEM
- Dulbecco’s modified Eagle’s Medium
- FBS
- fetal bovine serum
- HRP
- horseradish peroxidase
- PBS
- phosphate buffered saline
- BrdU
- bromodeoxyuridine
- TGF
- transforming growth factor
- PDGF
- platelet derived growth factor
- IFN
- interferon
- MDA
- malondialdehyde
Statistics from Altmetric.com
Hepatic fibrosis is a consequence of severe liver damage, which occurs in many chronic liver diseases as a forerunner to cirrhosis.1 ,2 It consists of an abnormal accumulation of extracellular matrix (ECM) proteins, particularly collagen.3 The development of cirrhosis is more common in men than women; the male:female ratio has been reported to range from 2.3:1 to 2.6:1.4 ,5 Although the liver is not a classic sex hormone target, livers in both men and women have been shown to contain high affinity, low capacity oestrogen receptors6 ,7 and respond to oestrogens by regulating liver function.8 ,9 Therefore, sex hormones may play a role in the progression from hepatic fibrosis to cirrhosis. However, the reasons why this occurs at a greater frequency in men suffering from chronic liver diseases is yet to be elucidated.
Stellate cells (fat storing cells, lipocytes, Ito cells) in the space of Disse have two known functions: primary storage of the body’s retinoids in intact liver lobules10 ,11 and the production of ECM components in the injured liver.12-14 When activated, these cells are transformed into myofibroblast-like cells which proliferate and express α smooth muscle actin (α-SMA), whereas both stellate cells and myofibroblasts express the intermediate filament, desmin.15 A loss of cellular retinoids occurs, and the cells synthesise large amounts of ECM components such as type I, III, and IV collagens, fibronectin, laminin, and proteoglycans. Therefore, the transformation of stellate cells into myofibroblast-like cells is now recognised as a critical step in hepatic fibrosis.
We recently found that oestradiol treatment resulted in reduced hepatic fibrosis in male rats in which hepatic fibrosis had been induced by a single dose of dimethylnitrosamine (DMN).16 This was associated with a reduced expression of type I procollagen and a tissue inhibitor of metalloproteinase 1 in the liver. Additionally, the deposition of type I and III collagens and hepatic collagen content were both reduced. In contrast, treatment with a neutralising antirat oestradiol antibody enhanced fibrogenesis in this DMN model. The suppressive effects of oestradiol on DMN induced hepatic fibrosis were also assessed in normal and castrated rats of both sexes.17 Thus, it is important to clarify whether oestradiol suppresses the expression of α-SMA, an activation marker of stellate cells, and restores the content of retinoids in experimental fibrotic livers induced in rats by treatment with either DMN18 or pig serum (PS).19 Low retinoid concentrations have been observed in various types of human liver disease20 as well as experimental fibrotic livers,21 suggesting that the hepatic concentration of retinoids, mainly retinyl palmitate (RP),11 may be a surrogate marker of hepatic fibrogenesis.21 ,22 The PS model for hepatic fibrosis, which shows neither hepatocellular damage nor regeneration,19 ,23 was used in this study. Additionally, the effects of oestradiol on myofibroblastic transformation of rat stellate cells in primary culture were assessed.
Materials and methods
IN VIVO MODEL
Preparation of animals
Male Wistar rats weighing 200 g were used for the DMN model and 120 g rats were used for the PS model. The rats were housed in air conditioned animal quarters with lighting from 0800 to 2000, with unrestricted access to a basal diet (CE-2; Nihon Clea, Tokyo, Japan) and water. As the experimental course for the DMN and PS models required two and 10 weeks, respectively, the lower weight animals were used for the latter model. Twenty five animals were used for the DMN model and 15 for the PS model (eight groups of five each) (fig 1).
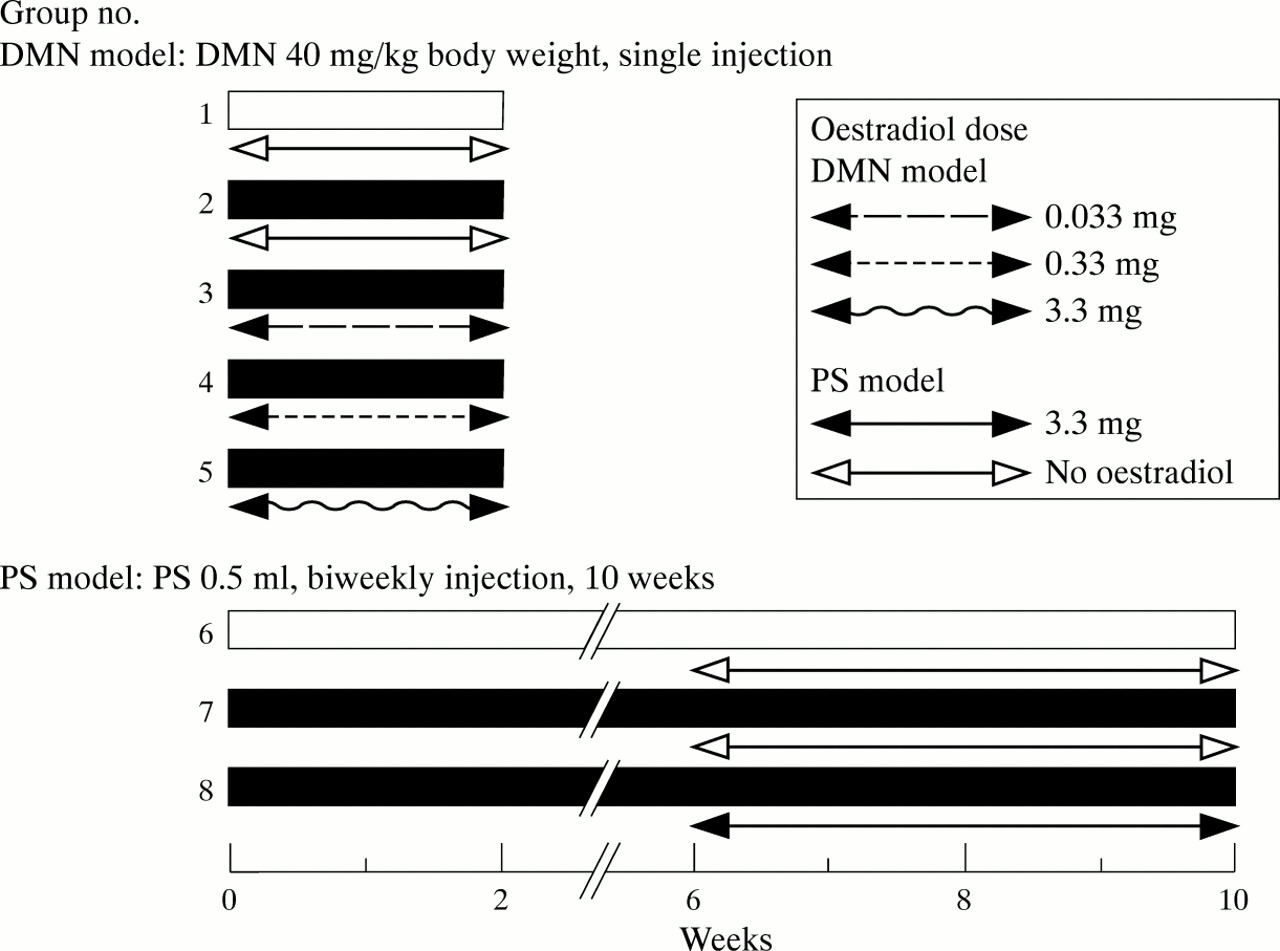
Experimental schedule in dimethylnitrosamine (DMN) and pig serum (PS) treated rats. Twenty five male Wistar rats were used for the DMN model and 15 for the PS model of hepatic fibrosis (eight groups of five each).
The DMN animals used in groups 2–5 were administered a single intraperitoneal injection of DMN (Sigma, St Louis, Missouri, USA) diluted with saline at a dose of 40 mg/kg body weight.18 ,24 Control animals for the DMN model in group 1 received a single injection of 0.5 ml of saline without DMN. The PS animals in groups 7 and 8 were administered intraperitoneal injections of PS (0.5 ml) twice per week for 10 weeks.19 The control animals for the PS model in group 6 received biweekly injections of saline (0.5 ml) for 10 weeks.
For the DMN model, the rats in groups 3, 4, and 5 received intraperitoneal injections of oestradiol valerate (Mochida Pharmaceutical Co., Tokyo, Japan) in olive oil at a dose of 0.033, 0.33, and 3.3 mg/kg/day, respectively, for two weeks after the single injection of DMN. For the PS model, the rats in group 8 received intraperitoneal oestradiol valerate in olive oil at a dose of 3.3 mg/kg/day for two weeks followed by a dose of 10 mg/kg twice per week for two weeks during the last four weeks of PS treatment. Animals in groups 1, 2, 6, and 7 received injections of olive oil without oestradiol.
All animals were anaesthetised using sodium pentobarbital (40 mg/kg, intraperitoneally) and killed after terminal exsanguination two weeks after DMN treatment in groups 1–5, and 10 weeks after PS treatment for groups 6–8. Blood samples were drawn from the inferior vena cava and used for radioimmunoassay of oestradiol. Liver tissue specimens were used for light microscopy and immunohistochemistry using anti-α-SMA and antidesmin antibodies. The remaining liver tissue was promptly removed and cut into small pieces; some were used for the direct determination of hepatic contents of collagen and RP, a main retinyl ester,11 while others were frozen in liquid nitrogen and stored at −80°C for RNA extraction. All animals used were treated humanely according to the Japanese National Guidelines.
Light microscopy and immunohistochemistry
For light microscopic examination, liver specimens were fixed overnight in phosphate buffered formaldehyde, embedded in paraffin wax, and stained with haematoxylin and eosin and Mallory azan. For immunohistochemical studies, tissue block sections were mounted on slides, deparaffinised in xylene, and rehydrated in alcohol. Endogenous peroxidase was blocked with a 1% hydrogen peroxide solution in methanol. Incubation with a monoclonal antibody directed against α-SMA (clone 1A4; Dako, Glostrup, Denmark; diluted 1/50) or desmin (Dako; diluted 1/25) for one hour at room temperature was preceded by digestion in a 0.1% trypsin solution in phosphate buffered saline (PBS) at 37°C for 30 minutes. The secondary antibody was a biotin conjugated rabbit antimouse IgG F(ab′)2 fragment (Dako; diluted 1/200) followed by incubation with an avidin-biotin complex (Vectastain ABC reagent, Vector Laboratories, Burlingame, California, USA). Reaction products were visualised using an ABC-peroxidase-diaminobenzidine method. A control for immunostaining specificity in which the primary antibody was substituted with non-immune mouse serum, was negative. Liver tissue sections were photographed using a light microscope (Carl Zeiss, Heidenheim, Germany) or a differential interference contrast (DIC) microscope (Axioskop, Carl Zeiss).
For the morphometric analysis of activated stellate cells in fibrotic livers, the mean value of α-SMA or desmin positive cells in five ocular fields per specimen was used as the percentage area at 40× magnification using an NIH image analysis system. As α-SMA was strongly stained in vascular smooth muscle cells and desmin was detected in perisinusoidal cells and vascular smooth muscle cells,15 the mean value in five control liver specimens from rats of groups 1 or 6 was subtracted from each experimental specimen and α-SMA or desmin positive cells were expressed as a percentage of the total area of the specimen.
Collagen content determination
Collagen was determined as previously reported.18 A portion of each liver was homogenised in 33 volumes (ml/g) of 0.5 M acetic acid at 4°C using a Polytron PCU-2 homogeniser (Kinematica, Switzerland), and the homogenate was disrupted by freeze thawing and sonication for two minutes. The fraction of insoluble collagen after the acid extraction, which was comprised of cross linked collagen, was then heated at 80°C for 60 minutes followed by conversion into soluble gelatin. The gelatin contents of the acid extract were assayed using the Sircol collagen assay kit (Biocolor, Belfast, Northern Ireland) according to the manufacturer’s directions, and the results were expressed as micrograms of collagen per 100 mg of dry weight liver.
Retinoid analysis
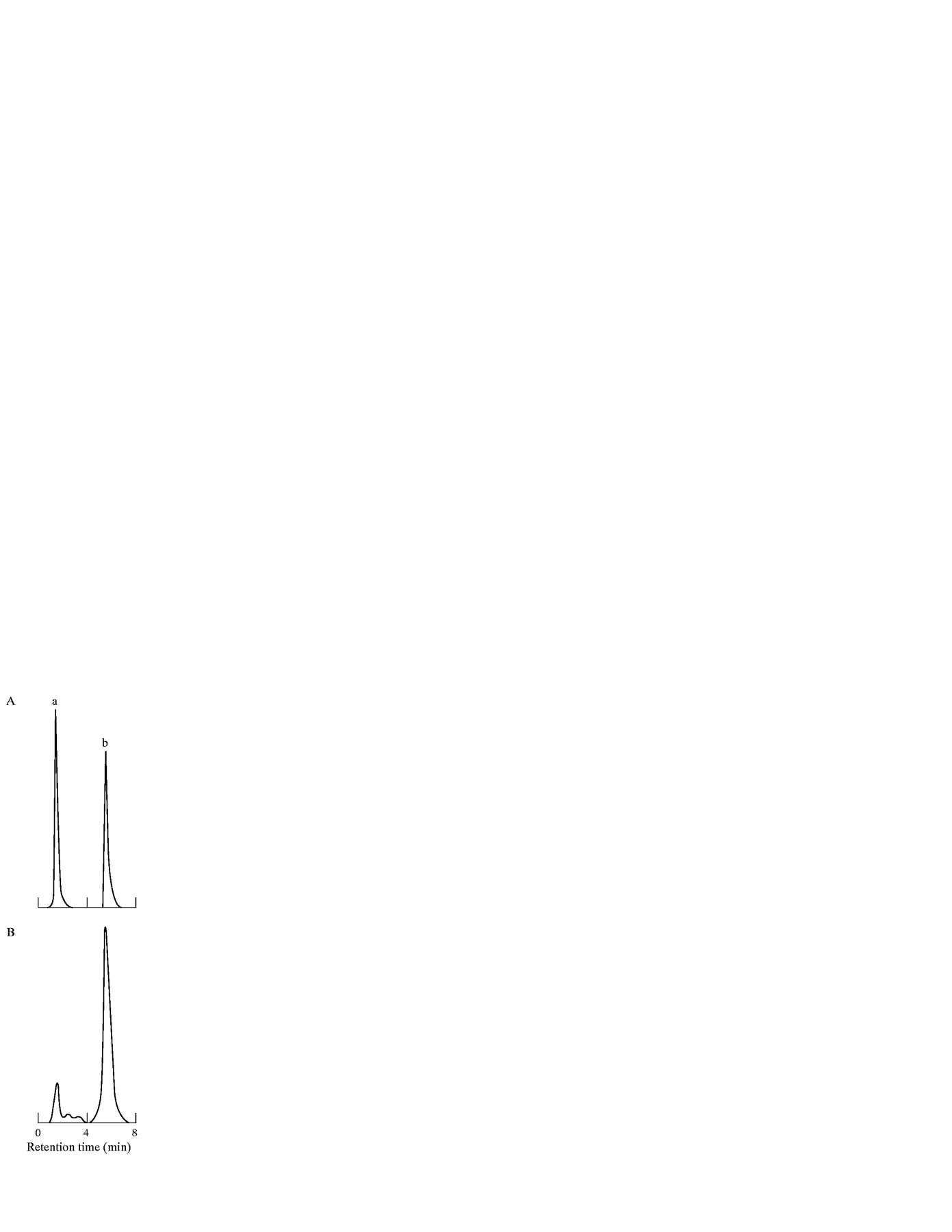
Retinoid analysis was performed as previously reported.21 Another portion of each liver was homogenised in 40 volumes (ml/g) of distilled water at 4°C using a Polytron PCU-2 homogeniser. For extraction, RP and retinyl acetate were added as the internal standard and the homogenate was mixed with two volumes of ethanol and n-hexane (containing 0.02% butylated hydroxytoluene). After shaking for five minutes, the mixture was centrifuged at 200 g to separate then-hexane layer. Then-hexane layer was removed and dried under a stream of N2. The residue was resuspended in a small volume of n-hexane and injected directly into a reverse phase high performance liquid chromatography (HPLC) column, TSKgel ODS-80Tm (Tosho, Tokyo, Japan) using an eluent composed of ethanol and distilled water (95:5) at a flow rate of 15 ml/h. Retinoids were detected by fluorescence spectorophotometry with an excitation wave length of 325 nm and an emission wave length of 470 nm. Hepatic RP content was quantified by comparing the peak areas of the extract samples with the peak area of authentic RP as an internal standard (peak b in fig 2), and corrected for differences in molar absorption coefficients as determined in standard solutions. For a series of 20 HPLC determinations, the efficiency of RP extraction was 78 (8)% with an intra-assay variability of 8 (4)%. The lowest detectable concentration of RP was 8.7 IU/injection (7 IU/g liver), evaluated as the concentration resulting in a response of 2 SD from the zero dose response.
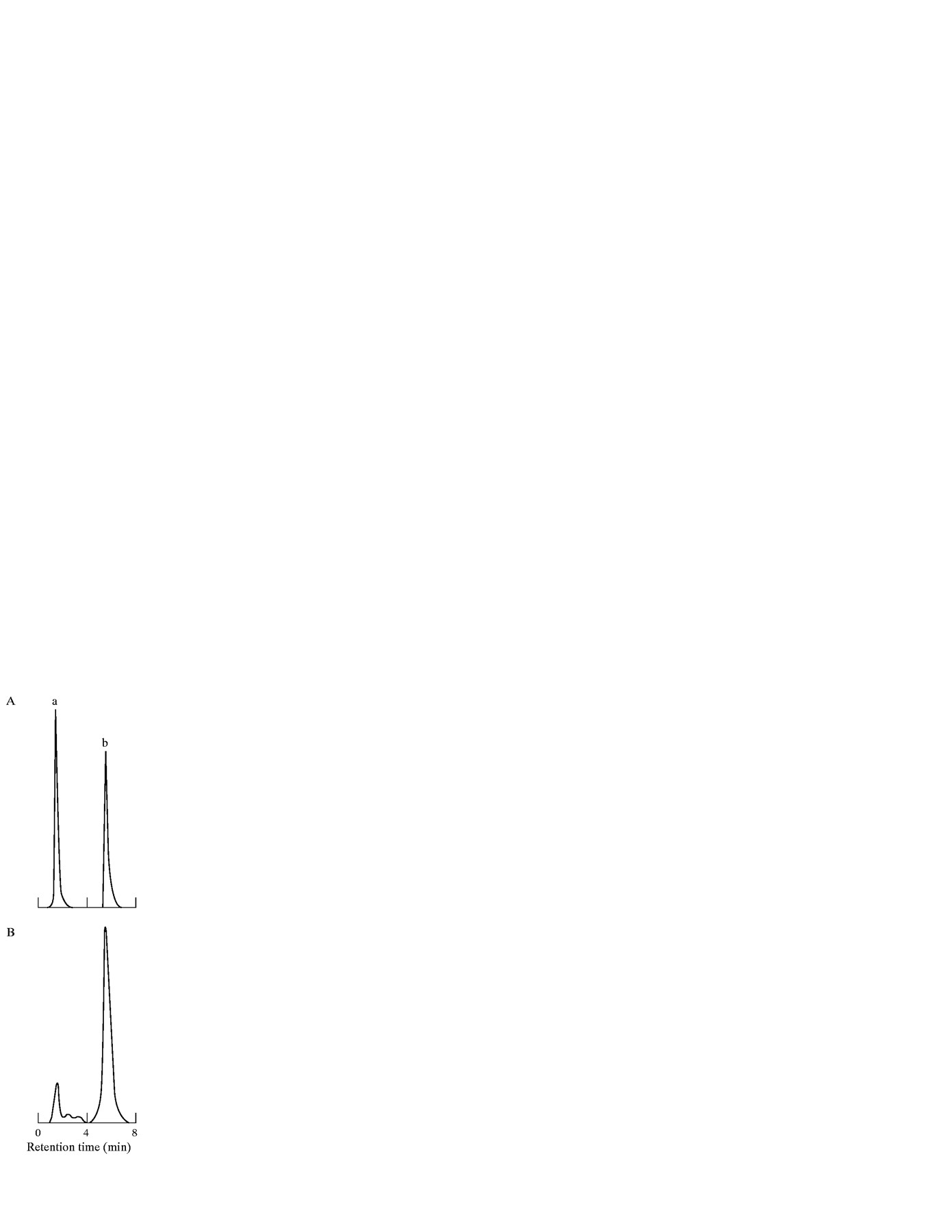
Representative HPLC chromatograms of (A) retinyl acetate and retinyl palmitate (RP) standards; and (B) a rat liver extract. Peaks: a, retinyl acetate (retention time, 1.9 minutes); b, RP, internal standard (retention time, 6.7 minutes).
RNA extraction and northern blot analysis
For total RNA extraction from liver tissue, 100 mg of the frozen liver tissue was homogenised in 2 ml of RNAzol B (Biotex Laboratories, Friendswood, Texas, USA) at 4°C using a Polytron PCU-2 homogeniser. Chloroform (200 μl) was added to the homogenate and the aqueous phase was collected after centrifugation at 12 000g at 4°C for 15 minutes. The RNA was precipitated with an equal volume of isopropanol and washed with 75% ethanol. RNA pellets were resuspended in diethylpyrocarbonate treated distilled water, and the amount of RNA was determined by spectrophotometric absorption at 260 nm. Samples of 10 μg of total RNA were electrophoresed under denaturing conditions, transferred to Hybond N+ filters (Amersham, UK), and fixed by ultraviolet cross linking. The filters were prehybrised and then hybridised with cDNA probes that were labelled with 32P-deoxcytidine triphosphate using a Random Prime DNA labelling kit (Boehringer Mannheim, Indianapolis, Indiana, USA).
The following cDNA probes were used for northern blots: a 3.55 kb EcoR1/EcoR1 fragment of clone NJ3 3.55 coding for pro-α2(I) collagen25; a 1.3 kb EcoR1/HindIII fragment of clone Hf934 coding for pro-α1(III) collagen26; and a 1.2 kb EcoR1/HindIII fragment of clone pHcGAP coding for glyceraldehyde-3-phosphate dehydrogenase (GAPDH).27 The probes were purchased from the American Type Culture Collection (Rockville, Maryland, USA). Hybridisation was performed overnight at 42°C in 50% formamide, 0.05 M sodium phosphate, 0.8 M NaCl, 1 mM EDTA, 0.2% sodium dodecyl sulphate (SDS), 2.5× Denhardt’s solution, and 250 μg/ml denatured salmon sperm DNA. Posthybridisation washes were performed in 2× standard saline citrate (1× SSC is 150 mM NaCl, 15 mM sodium citrate, pH 7.4) containing 0.1% SDS three times for 10 minutes at room temperature and also in 0.1× SSC containing 0.1% SDS three times for 20 minutes at 50°C. The filters were then exposed to Kodak XAR film (Eastman Kodak, Rochester, New York, USA) at −70°C. The filters were boiled in 0.1× SSC containing 0.1% SDS for 30 minutes to strip off the radioactive probes and rehybridised with another 32P-labelled cDNA probe in a similar manner. Densitomery tracings were recorded using a laser densitometer UltraScan XL (Pharmacia LKB, Uppsala, Sweden).
Radioimmunoassay of oestradiol
Serum oestradiol concentrations were measured radioimmunologically using a commercial kit (CIS Diagnostic, Tokyo, Japan). The lower limit of detection for the oestradiol assay system was 1.0 pg/ml.
IN VITRO STUDIES
Isolation and cultivation of stellate cells
Stellate cells were isolated from the livers of male Wistar rats (500–600 g) as described by Friedman et al.28 Briefly, the liver was perfused in situ through the portal vein with a 16 gauge cannula, first with Ca2+/Mg2+ free Krebs-Ringer (KR) solution at 37°C for 10 minutes at a flow rate of 10 ml/min, followed by 0.1% pronase E (Merck, Darmstadt, Germany) in KR solution for 10 minutes, and then with 0.3% collagenase (Wako, Osaka, Japan) in KR solution for 30 minutes. The digested liver was excised, minced with scissors, and incubated in KR solution containing 0.05% pronase E and 20 μg/ml DNAse (Boehringer Mannheim) for 30 minutes at pH 7.2. The resulting suspension was filtered through nylon mesh (150 μm in diameter). A stellate cell enriched fraction was obtained by centrifugation of the filtrate in an 8.2% Nycodenz (Nycomed, Oslo, Norway) solution at 1400g at 4°C for 20 minutes. The cells in the upper layer were washed by centrifugation at 450g at 4°C for 10 minutes and suspended in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS, Flow, McLean, Virginia, USA), 100 U/ml penicillin, 100 μg/ml streptomycin, and 1% l-glutamine. The purity of the isolated stellate cells was assessed by direct cell counting under a phase contrast microscope, by intrinsic vitamin A autofluorescence, and by immunocytochemistry using a monoclonal antibody against desmin (Dako; diluted 1/25); cell viability was examined by Trypan blue exclusion. Both cell purity and viability were in excess of 90%.
Cells were plated at a density of 5×105 cells per well in 1 ml of culture medium on uncoated plastic culture dishes (Nunc, Naperville, Illinois, USA). Culture medium was changed two days after plating to a culture medium containing β oestradiol (Wako) and was changed every other day thereafter. β Oestradiol was first prepared as an ethanolic stock solution (10−2 M) and then diluted in culture medium to the appropriate working solution (for example, 10× of the final concentration). Cells were maintained at 37°C in a 5% CO2 atmosphere and 100% humidity, and maintained in culture for 10 days.
Immunohistochemistry
Stellate cells which were initially cultured for two days in DMEM supplemented with 10% FBS were then maintained either in the presence or absence of oestradiol for an additional four days. Cells were then fixed with 0.5% paraformaldehyde for one hour at 4°C, washed twice with ice cold PBS, and incubated with 0.25% bovine testicular hyaluronidase for 30 minutes at 4°C. Cells were again washed twice with PBS at room temperature and incubated with a monoclonal antibody against α-SMA (Dako; diluted 1/50) for one hour at 37°C in a humid chamber. Cells were then washed twice with PBS and incubated with the secondary antibody, a biotin conjugated rabbit antimouse IgG F(ab′)2 fragment (Dako; diluted 1/200), and finally by the avidin-biotin complex (Vectastain ABC reagent, Vector Laboratories). Reaction products were visualised by an ABC-peroxidase-diaminobenzidine method.
ELISA analysis
Stellate cells cultured for two days were further incubated in the presence or absence of oestradiol for the indicated time period. The culture media were stored at −20°C, and the concentration of soluble type I collagen in these media was determined by a sensitive antigen enzyme linked immunosorbent assay (ELISA) method. Briefly, a murine monoclonal antibody to human type I collagen (Calbiochem, Cambridge, Massachusetts, USA) was pipetted at a 1/500 dilution in 100 μl of carbonate-bicarbonate buffer (pH 9.5) into Nunc-Immuno Module MaxiSorp 96 well plates. This solid phase was then allowed to absorb overnight at room temperature. Each plate was then aspirated and treated with PBS containing 0.5% non-fat milk (330 μl/well) overnight at 4°C. Following aspiration, the samples were diluted 1/1 with assay buffer (PBS with 0.05% Tween 20 and 0.2% non-fat milk) and subjected to a primary incubation for two hours at room temperature. The plates were washed five times with PBS containing 0.05% Tween 20 and then incubated with the second antibody, rabbit antihuman type I collagen (Chemicon, Temecula, California, USA) conjugated with horseradish peroxidase (HRP) using Pierce’s EZ-Link Plus activated peroxidase (Rockford, Illinois, USA) at a dilution of 1/500 in the assay buffer with 5% normal rabbit serum for two hours at room temperature. The plates were washed five times as described above and treated with 100 μl of 0.05 M citrate-phosphate buffer (pH 5.0) containing 1.0 mg/ml o-phenylene-diamine per well for 20 minutes at room temperature. The reaction was stopped by the addition of 1 N sulphuric acid. The plates were read in an Auto Reader (MTP-120, Corona Electric, Katsuta, Japan) using a 492 nm filter with a reference at 630 nm, after blanking the machine with a reagent blank plate. Sample concentrations were determined from a plot of optical density versus concentration of type I collagen standard over a range of 0–100 μg/ml.
Cell proliferation
Using the Biotrak cell proliferation ELISA system (Amersham, Little Chalfont, UK), DNA synthesis was measured by incorporation of the pyrimidine analogue bromodeoxyuridine (BrdU) into the DNA of proliferating cells.29 Stellate cells cultured in 96 well plates for four days were further incubated in the presence or absence of oestradiol for 24 hours and were labelled with 10 μM BrdU for another 24 hours. After removing the culture medium, the cells were fixed and the incorporated BrdU was detected by the subsequent substrate reaction according to the manufacturer’s recommended protocol.
Western blot analysis of α-SMA
Stellate cells were cultured in 1 ml of culture medium with or without oestradiol for the indicated time period, and were then washed twice with ice cold PBS and lysed directly in SDS loading buffer (50 mM Tris-HCl, pH 6.8, 100 mM dithiothreitol, 2% SDS, 0.1% bromophenol blue, and 10% glycerol). The samples were boiled for 10 minutes and applied to a standard 12% SDS-polyacrylamide protein gel. After electrophoresis, protein transfer was performed onto Hybond-ECL (Amersham, Arlingtone Heights, Illinois, USA) using a semi-dry blotting apparatus. The membrane was treated first with 10% non-fat milk in PBS and next with the monoclonal anti-α-SMA antibody (Dako; diluted 1/500) for two hours at room temperature. After vigorous washing, the membrane was then incubated with HRP conjugated goat antimouse IgG (Amersham; diluted 1/1000) for one hour at room temperature. Immunoreactive bands were visualised using the ECL (enhanced chemiluminescence) western blotting detection system kit (Amersham) according to the manufacturer’s recommended protocol.
STATISTICAL ANALYSIS
Data are presented as means (SD) unless otherwise indicated. Means were compared with the Wilcoxon rank sum test. A p value of less than 0.05 was considered significant.
Results
IN VIVO EFFECT OF OESTRADIOL ON THE ACTIVATION OF RAT HEPATIC STELLATE CELLS
As α-SMA is an activation marker of rat hepatic stellate cells and desmin is reported to be stained in stellate cells and myofibroblasts as well as smooth muscle cells,15 the expression of these proteins was analysed by immunohistochemistry. In control rat livers in groups 1 and 6, desmin was detected in the sinusoids in a fibre-like pattern and weak staining for it was observed in vascular smooth muscle cells; α-SMA was strongly stained in vascular smooth muscle cells, but not in the sinusoids (data not shown).
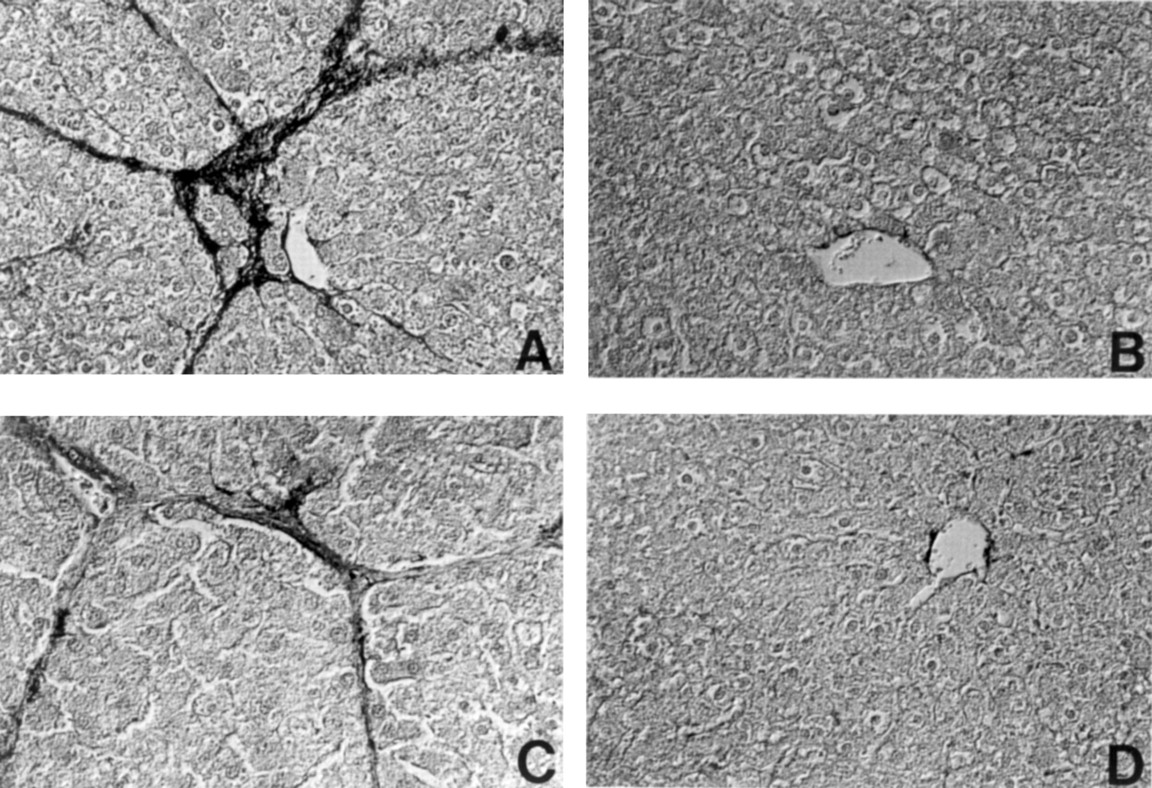
In the DMN model, a single intraperitoneal administration of DMN induced fibrotic changes in the liver in a dose dependent manner, mainly around the central and portal veins, 14 days after treatment.18 Immunohistological analysis performed on liver sections from rats treated with 40 mg/kg DMN showed a considerable number of high percentages of 8.3 (1.0)% and 6.2 (1.3)% of α-SMA and desmin positive cells, respectively, within centrilobular and periportal fibrotic bands (figs 3A, 4A, and5).
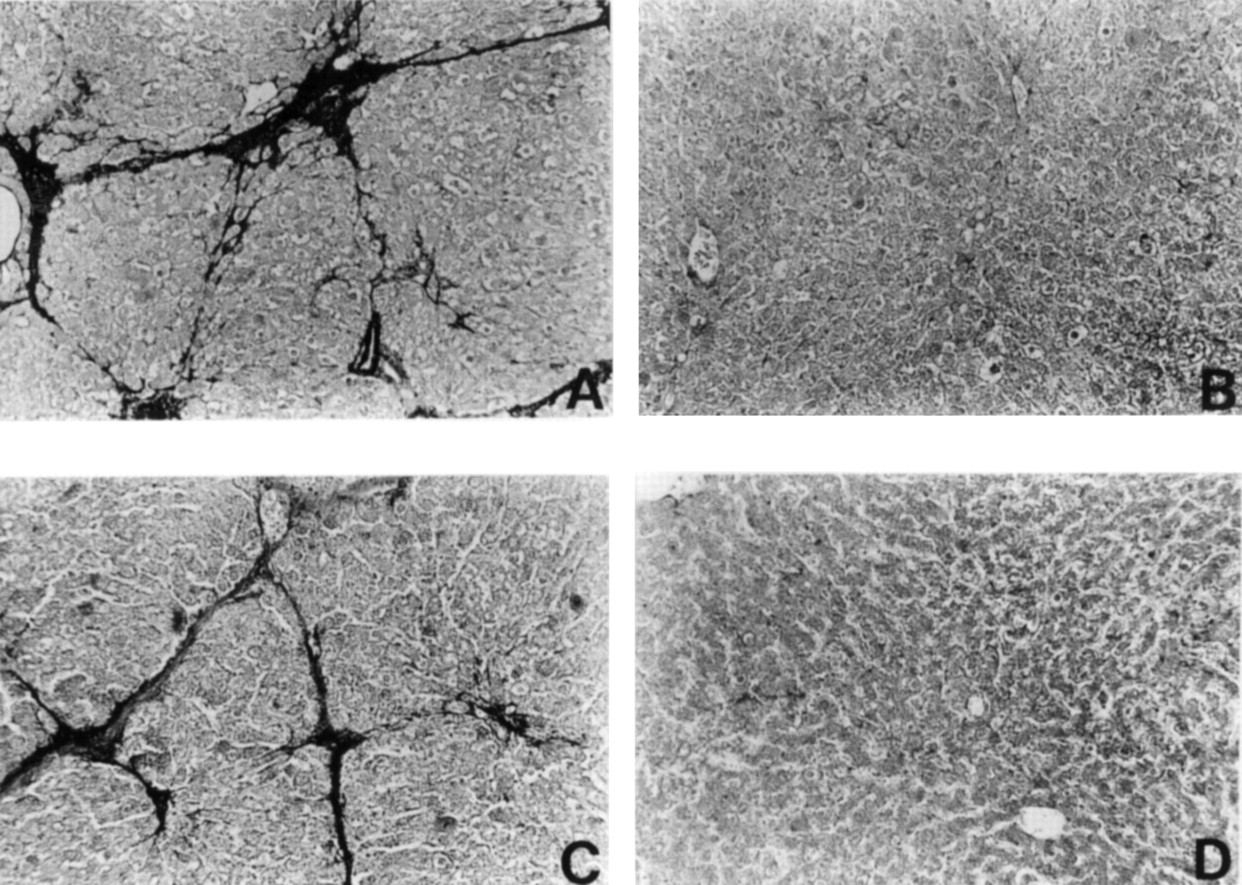
Reaction of representative liver sections with antibody to α smooth muscle actin (α-SMA). (A) Group 2; (B) Group 5; (C) Group 7; (D) Group 8. Original magnification ×60.

Reaction of representative liver sections with an antibody to desmin. (A) Group 2; (B) Group 5; (C) Group 7; (D) Group 8. Liver sections were photographed using a differential interference contrast microscope. Original magnification ×90.

Percentage areas of α smooth muscle actin (α-SMA) and desmin positive cells after treatment with varying doses of oestradiol in the dimethylnitrosamine (DMN) and pig serum (PS) models. The mean value of α-SMA or desmin positive cells in six ocular fields (magnification ×40) per specimen was assessed as percentage area of α-SMA or desmin positive cells in six ocular fields at 40× magnification with five control rat livers. The area was measured using microcomputer based image analysis. *p<0.05 compared with rats treated with the same reagent (DMN or PS) but not given oestradiol.
In the PS model, connective tissue septa interlinked central and portal veins, and pseudolobuli were produced without hepatocytic degeneration, necrosis, or regeneration by 6–10 weeks of PS administration. Aggregates of activated stellate cells within fibrotic bands were labelled with antibodies against α-SMA and desmin (figs 3C and 4C). The percentage areas of α-SMA and desmin positive cells were 6.3 (0.9)% and 4.9 (1.1)%, respectively. The difference between the percentage areas of α-SMA and desmin positive cells in both the models of hepatic fibrosis suggests that α-SMA positive cells within the fibrous septa might be derived from progenitor cells other than stellate cells, for example, resident myofibroblasts. In contrast, the coadministration of oestradiol notably reduced the areas of α-SMA and desmin positive cells in the livers of rats treated with DMN (figs 3B,4B, and 5) and PS (figs 3D, 4D, and 5). Furthermore, oestradiol at a dose of 0.33 and 3.3 mg/kg significantly reduced the percentage areas of α-SMA and desmin positive cells in DMN induced fibrotic livers in a dose dependent manner (fig 5).
The decreased area of α-SMA positive stellate cells was confirmed by findings of lowered hepatic collagen content and restored hepatic RP content after oestradiol treatment (fig 6). Fibrotic livers in the DMN and PS models had 2.2-fold and 1.4-fold higher collagen concentrations, respectively, than control livers. Oestradiol treatment resulted in reduced collagen content in a dose dependent manner in the DMN model and reduced concentrations to control values in the PS model (fig 6). Furthermore, the RP content of the DMN and PS induced fibrotic livers decreased significantly by 47% and 49%, respectively, compared with control values, while oestradiol restored the hepatic RP contents to control concentrations (fig 6). Even the lowest dose of oestradiol of 0.033 mg/kg restored the hepatic RP content in DMN treated rats (fig6).

Effects of oestradiol on hepatic collagen and retinyl palmitate (RP) content in rats treated with dimethylnitrosamine (DMN) or pig serum (PS). All results are expressed as percentages (mean (SD)) of control values: in the DMN model, 765 (267) μg collagen/100 mg liver and 206 (34) IU RP/g liver; in the PS model, 796 (231) μg collagen/100 mg liver and 266 (36) IU RP/g liver. *p<0.05 compared with controls; †p<0.05 compared with rats treated with the same reagent (DMN or PS) but not given oestradiol.
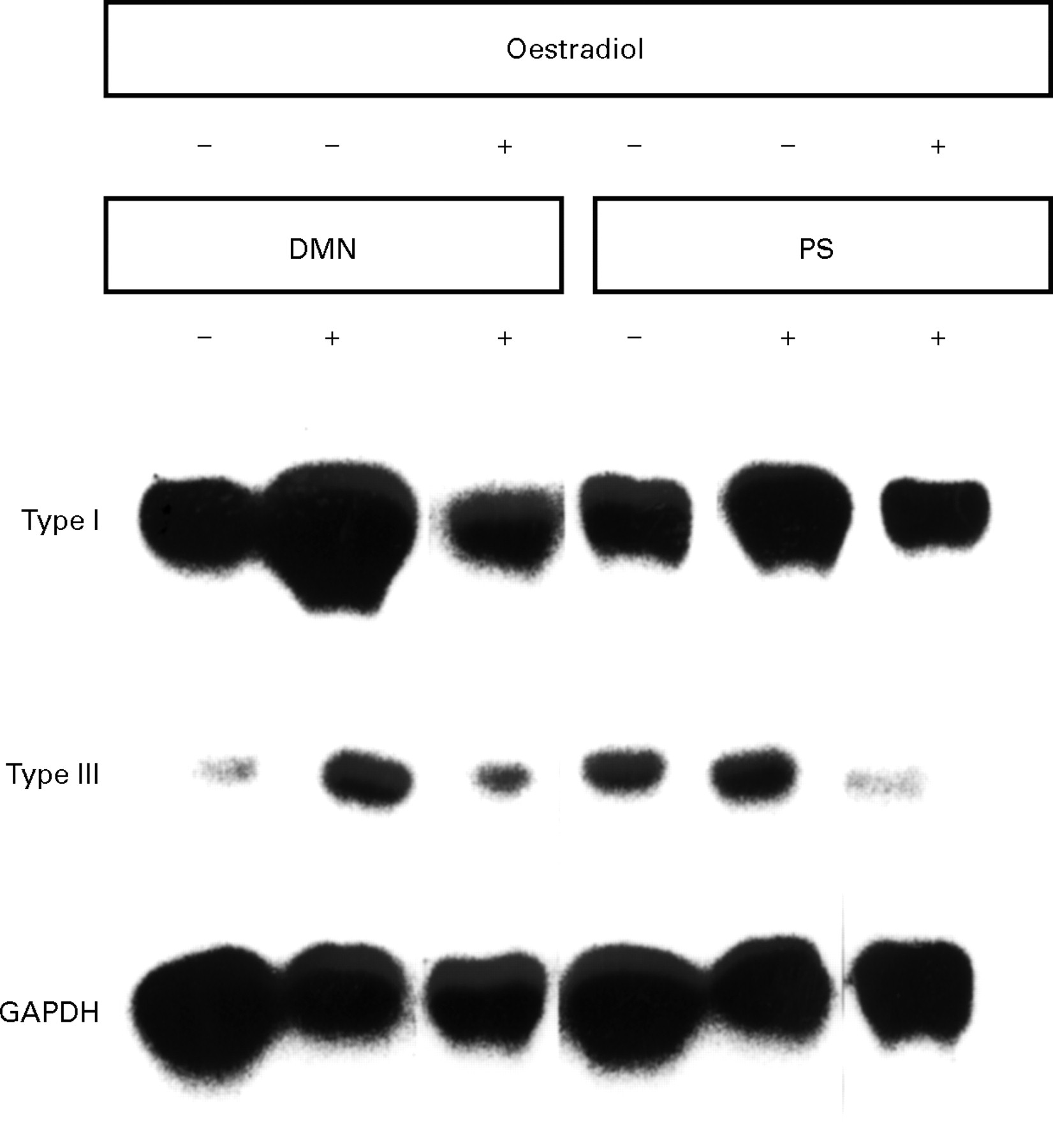
Northern blot analysis was performed with extracts from livers of DMN and PS treated rats either in the presence or absence of oestradiol from three separate experiments. As shown in the representative figure (fig 7), mRNA expression levels of procollagen types I and III were notably increased in the fibrotic livers from the DMN and PS models, as compared with control livers. Extracts from the oestradiol treated groups showed lower mRNA levels of these procollagens than did the extracts from the group treated with DMN or PS alone. GAPDH mRNA levels were equivalent in all extracts (fig 7).
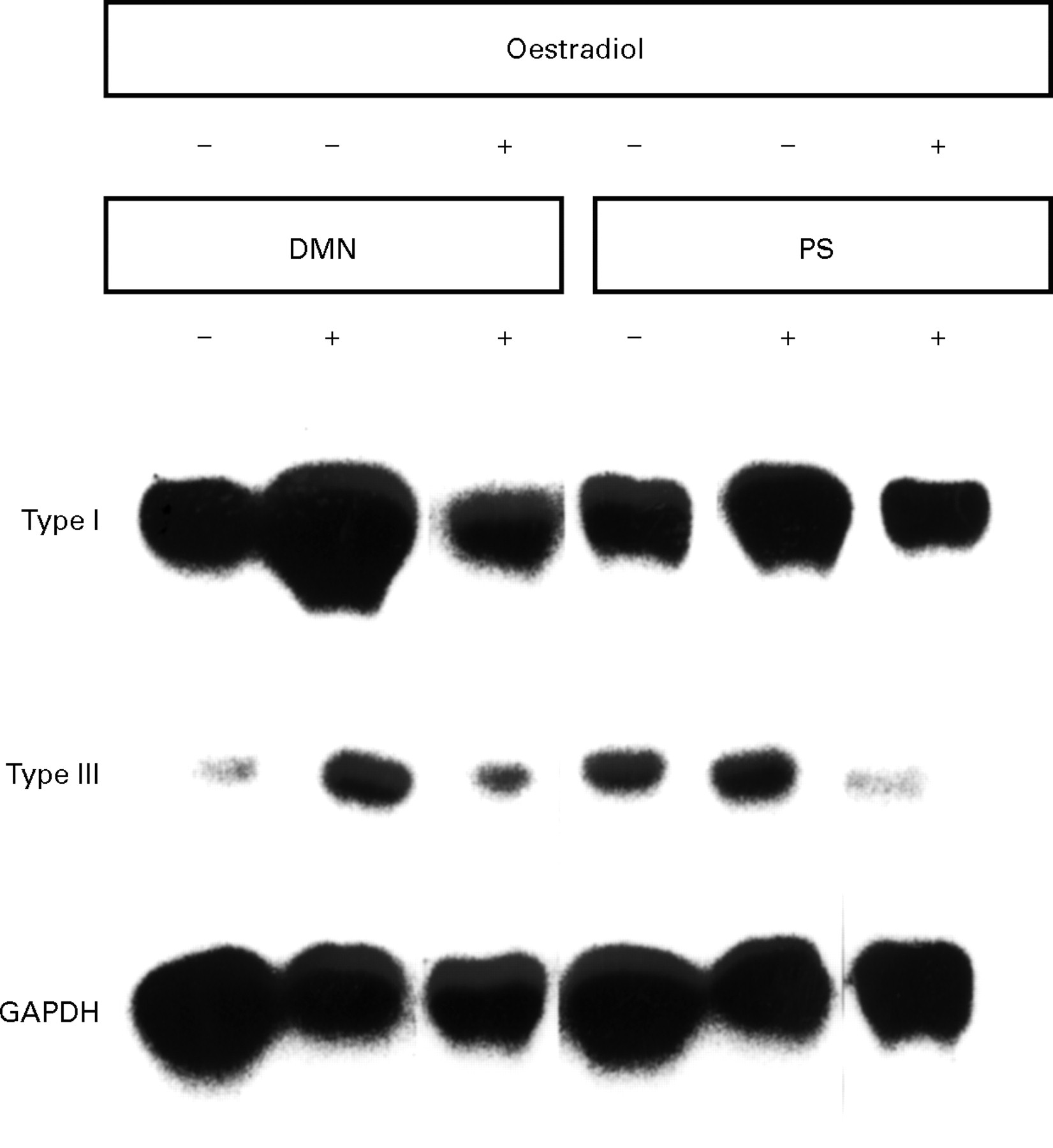
Northern blot analysis undertaken with extracts from livers of control rats or dimethylnitrosamine (DMN) and pig serum (PS) treated rats in the presence or absence of oestradiol from three separate experiments.
Table 1 shows the mean (SD) concentrations of serum oestradiol in the DMN and PS models with or without oestradiol treatment. No significant difference was observed among the DMN and PS models without oestradiol treatment and the controls. In the DMN model, oestradiol treatment at a dose of 0.033, 0.33, or 3.3 mg/kg/day for two weeks significantly induced serum oestradiol concentrations in a dose dependent manner, comparable to concentrations observed during the late follicular phase of the normal menstrual cycle, early, and late gestation phase, respectively. In the PS model, the oestradiol concentrations after oestradiol treatment increased to concentrations comparable to late gestation related concentrations.
Effect of oestradiol on serum oestradiol concentrations in dimethylnitrosamine (DMN) and pig serum (PS) treated rats
IN VITRO EFFECT OF OESTRADIOL ON THE ACTIVATION OF RAT HEPATIC STELLATE CELLS
Stellate cells underwent transformation to myofibroblast-like cells with enlarged cell bodies containing less lipid particles and exhibiting immunoreactive α-SMA within five days after culture on uncoated plastic dishes in DMEM supplemented with 10% FBS. Oestradiol (10−8–10−6 M) was added to stellate cell cultures two days after cell plating and incubation was continued for up to eight days. Six days after cell plating, most of the cells cultured in DMEM/10% FBS were well spread and showed α-SMA microfilaments (fig 8A). Supplementation of the medium with oestradiol (10−6 M) inhibited cell spreading and the degree of immunocytochemical staining of α-SMA on day 6 (fig 8B). This decrease in α-SMA staining was reproduced in western blot analysis for α-SMA (fig 9). Expression of α-SMA increased during time in culture, and was reduced at all time points when stellate cells were cultured in the presence of oestradiol (10−7 M) (fig 9A). The inhibitory effect of oestradiol (10−8–10−6 M) on α-SMA expression occurred in a dose dependent manner (fig 9B). Furthermore, such inhibitory oestradiol effects were confirmed by ELISA analysis of soluble type I collagen in the culture medium (table 2) and BrdU incorporation assays (fig 10). The amount of newly synthesised type I collagen in DMEM/10% FBS on day 10 was 1.8-fold greater than on day 5. In contrast, supplementation of the medium with oestradiol (10−8–10−6 M) inhibited this increase in new collagen synthesis in a dose dependent manner (table 2). The BrdU incorporation assay revealed that DNA synthesis in stellate cells was also suppressed by the presence of oestradiol (10−8–10−6 M) in a dose dependent manner (fig 10).

Reaction of cultured rat hepatic stellate cells with antibody to α smooth muscle actin (α-SMA). Cells were incubated in the (A) absence or (B) presence of oestradiol (10−6 M) for four days. Original magnification ×200.

Effect of oestradiol on α smooth muscle actin (α-SMA) expression in cultured rat hepatic stellate cells. (A) Cells cultured for an additional three (day 5) or eight (day 10) days in the presence (+) or absence (−) of oestradiol (10−7 M). (B) Cells cultured for an additional eight days in the presence of oestradiol (10−8–10−6 M).
Effect of oestradiol on type I collagen production in cultured rat hepatic stellate cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of oestradiol on DNA synthesis in cultured rat hepatic stellate cells. Cells were incubated in the presence or absence of oestradiol (10−8–10−6 M) for 24 hours before being labelled with bromodeoxyuridine (BrdU) for 24 hours. Results expressed as percentages (SD) (n=4) of control value. *p<0.05 compared with controls.
Discussion
Cell proliferation, α-SMA expression, retinoid disappearance, and the formation of collagens and other ECM materials are characteristics of the activated phenotype of hepatic stellate cells. These changes have been postulated to underlie the pathogenesis of hepatic fibrosis. In the present study we found that oestradiol at physiological doses, which induce concentrations comparable to gestation related concentrations, dose dependently suppressed the development of hepatic fibrosis in both the DMN and PS models with increased hepatic RP content, reduced collagen content, lower areas of stellate cells positive for α-SMA and desmin, and lower procollagen types I and III mRNA levels in the liver. Additionally, in cultured rat stellate cells, oestradiol inhibited type I collagen production, α-SMA expression, cell spreading, and DNA synthesis. These findings suggest that oestradiol is a potent inhibitor of stellate cell transformation, and thus, the antifibrogenic role of oestradiol in the liver may be one reason for the sex associated differences in the progression from hepatic fibrosis to cirrhosis.
Although desmin is generally characteristic of smooth muscle cells, it has been shown that desmin positive cells, found in perisinusoidal cells of normal livers and in fibrotic septa of cirrhotic livers, represent resident hepatic stellate cells in the former case, and activated stellate cells which transformed into myofibroblastlike cells, in the latter case15; furthermore, stellate cells are identified as perisinusoidal desmin positive, α-SMA-negative cells.30 The findings are consistent with the present data, which show that in control rat livers, perisinusoidal cells were positive for desmin, but not for α-SMA (data not shown), whereas in the fibrotic livers induced by DMN or PS α-SMA and desmin positive cells were located in the pericentral and periportal areas along fibrotic bands. In the present study, however, a difference between the percentage areas of α-SMA and desmin positive cells in both the DMN and PS models was observed: the former areas were larger than the latter areas. This suggests that α-SMA positive cells within the fibrous septa might be derived from progenitor cells other than stellate cells—for example, resident myofibroblasts. Additionally, it should be noted that the increased area taken up by α-SMA or desmin positive cells does not necessarily reflect hyperplasia but could reflect hypertrophy of stellate cells.
Hepatic fibrogenesis is often associated with hepatocellular necrosis and inflammation accompanied by the repair processes.31Stellate cells are regarded as the primary target cells for inflammatory stimuli in the injured liver.32 When activated by inflammatory stimuli, stellate cells are transformed into myofibroblast- like cells. DMN, which was used to induce fibrosis, is toxic to hepatocytes and can induce coagulation necrosis in hepatocytes predominantly in the central and periportal regions of the lobule.33 Inflammatory cells contribute further to the fibrogenic response to hepatotoxin via the release of cytokines, such as transforming growth factor β1 (TGF-β1) and platelet derived growth factor (PDGF). It has been shown that TGF-β1 and PDGF further activate cultured stellate cells,34 ,35 although stellate cells cultured on uncoated plastic dishes are already activated. As these growth factors are produced by infiltrating inflammatory cells, Kupffer cells and sinusoidal endothelial cells, they might act as paracrine mediators which trigger the transformation of stellate cells in vivo. In the second fibrosis model, PS is antigenic and is taken up by macrophages/Kupffer cells within the sinusoids.36Therefore, antigen and antibodies (immunoglobulins) can be detected in the sinusoids after several weeks,37 and stimulated Kupffer cells have the ability to activate stellate cells.38 The data presented here show that oestradiol is a potent inhibitor of stellate cell activation, playing an antifibrogenic role in regulating hepatic collagen formation, hepatic retinoid disappearance, and stellate cell proliferation. Interestingly, oestradiol had the ability to inhibit α-SMA expression in both the DMN and PS induced fibrotic livers and in cultured stellate cells in a dose dependent manner.
The mechanism by which oestradiol inhibits stellate cell activation is not clear. However, oestradiol administration has been shown to activate an extrathymic pathway of T cell differentiation in the liver,39 and to increase the activity of the interferon (IFN) γ promoter in lymphoid cells.40 IFN-γ is an potent cytokine with immunomodulatory and antiproliferative proterties. It has also been shown to have inhibitory effects on ECM production in in vivo models of hepatic schistosomiasis41 and carbon tetrachloride42 and DMN induced hepatic fibrosis,43 as well as on the activation of stellate cells in culture.44 In addition, evidence now exists that lipid peroxidation45 and acetaldehyde46 share a common mechanism in stimulating collagen gene expression, and that oestradiol and its derivatives (2-hydroxyoestradiol) are strong endogenous antioxidants that inhibit serum and liver lipid peroxide concentrations.47 Our preliminary study has shown that oestradiol inhibits lipid peroxidation in rat liver mitochondrial membranes induced by adenosine 5′-diphosphate (ADP) and FeSO4.48 Recently, Lee et al have reported that Ito cells are activated by the generation of free radicals with ascorbate/FeSO4 and malondialdehyde (MDA), a product of lipid peroxidation, and that stellate cell activation by type I collagen is blocked by antioxidants.49 These findings suggest that oestradiol may exert its suppressive effect on hepatic fibrosis, at least in part, through the stimulation of IFN-γ and/or the inhibition of lipid peroxidation.
There are also several reports that oestradiol treatment in vivo significantly lowered total collagen in animal aortas in comparison with testosterone treatment,50 and that oestradiol preferentially inhibited production of type III procollagen while not affecting the production of type I procollagen in cultured aortic smooth muscle cells.51 It should be noted that oestradiol plays an important role during the menstrual cycle and gestational period in normal women. Judging from these findings and the present data that oestradiol suppresses the development of hepatic fibrosis in males, the antifibrogenic role of oestradiol in the liver may be one reason for the preponderance of men suffering from chronic liver diseases.
Acknowledgments
The authors thank Dr M Shono for his help regarding photomicroscopic techniques. This work was supported in part by Grant-in-Aid for Scientific Research No. 06670559 from the Ministry of Education, Science and Culture of Japan.
Abbreviations
- SMA
- smooth muscle actin
- ECM
- extracellular matrix
- DMN
- dimethylnitrosamine
- PS
- pig serum
- RP
- reinyl palmitate
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- SDS
- sodium dodecyl sulphate
- DMEM
- Dulbecco’s modified Eagle’s Medium
- FBS
- fetal bovine serum
- HRP
- horseradish peroxidase
- PBS
- phosphate buffered saline
- BrdU
- bromodeoxyuridine
- TGF
- transforming growth factor
- PDGF
- platelet derived growth factor
- IFN
- interferon
- MDA
- malondialdehyde