Article Text

Abstract
BACKGROUND Omeprazole has a greater intragastric pH elevating effect in Helicobacter pylori positive than negative subjects. Ammonia production byH pylori has been suggested as a probable mechanism.
AIMS To assess the effect ofH pylori status on gastric acid secretion during omeprazole treatment, and to examine the possible role of ammonia neutralisation of intragastric acid in increased omeprazole efficacy in infected subjects.
METHODS TwentyH pylori positive and 12H pylori negative healthy volunteers were examined before and six to eight weeks after commencing omeprazole 40 mg/day. On both occasions plasma gastrin and acid output were measured basally and in response to increasing doses of gastrin 17 (G-17). Gastric juice ammonium concentrations were also measured.
RESULTS Prior to omeprazole, measurements were similar in the H pyloripositive and negative subjects. During omeprazole, median basal intragastric pH was higher in the H pyloripositive (7.95) versus negative (3.75) subjects (p<0.002). During omeprazole basal, submaximal (180 pmol/kg/h G-17), and maximal acid outputs (800 pmol/kg/h G-17) were lower in H pylori positive subjects (0.0, 3.6, 6.0 mmol/h respectively) versus negative subjects (0.3, 14.2, 18.6 mmol/h) (p<0.03 for each). This effect was not explained by neutralisation by ammonia.
CONCLUSION The presence ofH pylori infection leads to a more profound suppression of acid secretion during omeprazole treatment. The effect cannot be explained by neutralisation of intragastric acid by bacterial ammonia production and its precise mechanism has to be explained.
- omeprazole
- Helicobacter pylori
- ammonia
- acid secretion
Abbreviations
- BAO
- basal acid output
- ECL
- enterochromaffin-like
- G-17
- gastrin 17
- GORD
- gastro-oesophageal reflux disease
- MAO
- maximal acid output
- PPI
- proton pump inhibitor
Statistics from Altmetric.com
Omeprazole, a substituted benzimidazole, non-competitive inhibitor of the gastric proton pump,1 ,2 has become one of the world’s most frequently prescribed medications.3Early studies in duodenal ulcer patients4 and healthy volunteers5 showed its efficacy in producing profound suppression of acid secretion. This ability has been utilised with great success in a wide spectrum of acid related disorders.6-9
After the introduction of omeprazole, H pyloriwas recognised as a highly prevalent infectious agent of the gastric mucosa in both dyspeptic patients10-12 and asymptomatic healthy individuals.13 ,14 Omeprazole has been shown to exert effects on H pylori 15 ,16 and the associated gastritis.17-20
The presence of H pylori infection may also exert effects on the actions of omeprazole. Four studies have shown that intragastric pH during omeprazole treatment is higher inH pylori infected subjects than inH pylori negative or eradicated subjects.21-24 The investigators in these studies have concluded that the greater elevation of pH on omeprazole in the presence of H pylori is mainly due to production of ammonia by its urease enzyme,25 neutralising intragastric acid.
The aims of this study were: (1) to assess the effect ofH pylori status on gastric acid secretion, as opposed to intragastric pH, during proton pump inhibitor (PPI) treatment; and (2) to assess the contribution of H pylori ammonia production to any effects observed. Our findings show that the presence of H pylori leads to notably greater suppression of basal, submaximal, and maximal acid secretion during PPI treatment. They also show that ammonia production by H pylori and, indeed, neutralisation from any other source, cannot explain these observations.
Materials and methods
SUBJECTS STUDIED
Twenty H pylori negative healthy volunteers (10 men, five smokers) and 12 H pyloripositive healthy volunteers (four men, four smokers) were studied. The mean weight and age of the H pylori negative volunteers were 75.9 kg and 27.9 years; those of the H pylori positive volunteers were 71.4 kg and 29.5 years. None of these volunteers were taking any medication, other than oral contraceptives. None reported any major gastrointestinal symptoms.
H pylori status was determined using the14C-urea breath test. This test has been validated in our unit: using a cut off value of 30 (kg % dose/mmol CO2) for the 20 minute result it has a sensitivity of 98% and a specificity of 100%.26
METHODS
Basal gastrin concentration, basal intragastric pH, and basal acid output were measured in all subjects. Acid output was then measured in response to submaximal and maximal doses of gastrin 17 (G-17). Following this, the subjects took an eight week course of omeprazole 40 mg each morning (at 0900 hours) (Astra Hassle, Molndal, Sweden) with weekly reminder telephone calls and fortnightly tablet counts being carried out. During the last two weeks of this course, the gastrin and acid secretory studies were repeated 24 hours after the previous dose of omeprazole.
For the gastric secretory studies, all subjects reported at 0900 after a 12 hour fast. A 16F nasogastric tube (Andersen Inc., New York, USA) was passed and its position in the dependent part of the stomach was checked using the water recovery test.27 After the stomach was emptied, intermittent suction was applied using an intermittent suction unit (Ohmeda, Columbia, Maryland, USA), which applies suction for 20 seconds in each 32 second cycle. A 30 minute basal acid collection was obtained, then sequential 30 minute collections were made during infusions of G-17 at doses of 7, 20, 60, 180, and 800 pmol/kg/h. Blood samples were collected each morning for gastrin determination, both basally and at the end of each infusion period. The plasma was stored at −20°C. A gastric juice sample was taken at the end of both the basal and the peak G-17 infusion periods for later ammonium measurement. These gastric juice samples were stored at −70°C. Basal samples at both time points were unavailable for two of the H pylori negative subjects and one of the H pylori positive subjects, maximal samples for six of the H pylori negative subjects and three of the H pylori positive subjects. The pH and volume of each acid collection was noted and its hydrogen ion concentration was measured by titration with 0.1 M sodium hydroxide to pH 7.0 using an autotitrator (Radiometer ETS 822, Copenhagen, Denmark).
G-17 was purchased from Peninsula Laboratories (Belmont, California, USA) as aliquots of freeze dried lyophilised powder. Subsequent preparation was performed under sterile conditions by the Western Infirmary Pharmacy Department. Each aliquot was dissolved in a small volume of ammonium bicarbonate, then made up into a stock solution. Vials containing 100 μg of G-17 were prepared and stored at −20°C until the day of the study. For each study, the content of the vial was further diluted in 0.9% sodium chloride solution containing 1% human serum albumin (Scottish National Blood Transfusion Service, Law Hospital, Carluke, Scotland, UK).
Plasma gastrin levels were measured by radioimmunoassay using antiserum R98, which has a sensitivity of 5 ng/l and detects both sulphated and unsulphated forms of G-17 and G-34 with equal affinity.28
Before analysis for ammonium concentration, gastric juice samples were centrifuged at 3000 g for 10 minutes to remove mucus. The concentration of ammonium was measured in the supernatant after dilution in 0.2 mol/l phosphate buffer pH 7.4 using a previously validated enzymatic method (Sigma Chemicals) adapted for the Cobas Mira (Roche, Welwyn Garden City, UK).29 The interassay coefficient of variation ranged from 8.5% at 2.3 mmol/l gastric juice ammonium to 1.9% at 4.7 mmol/l, while the detection limit was 30 μmol/l. Gastric juice samples were diluted with phosphate buffer prior to analysis to prevent low gastric juice pH interfering with the enzyme used in the assay.29
STATISTICS
Statistical analysis was performed using a two tailed Mann-Whitney U test for unpaired data and a two tailed Wilcoxon sum rank test for paired data. A p value of less than 0.05 was considered significant. The healthy volunteers were recruited by advertising in the hospital’s catchment area. The study was approved by the West of Glasgow Hospitals University NHS Trust Ethics Committee and all subjects gave written, informed consent.
Results
BASAL INTRAGASTRIC pH
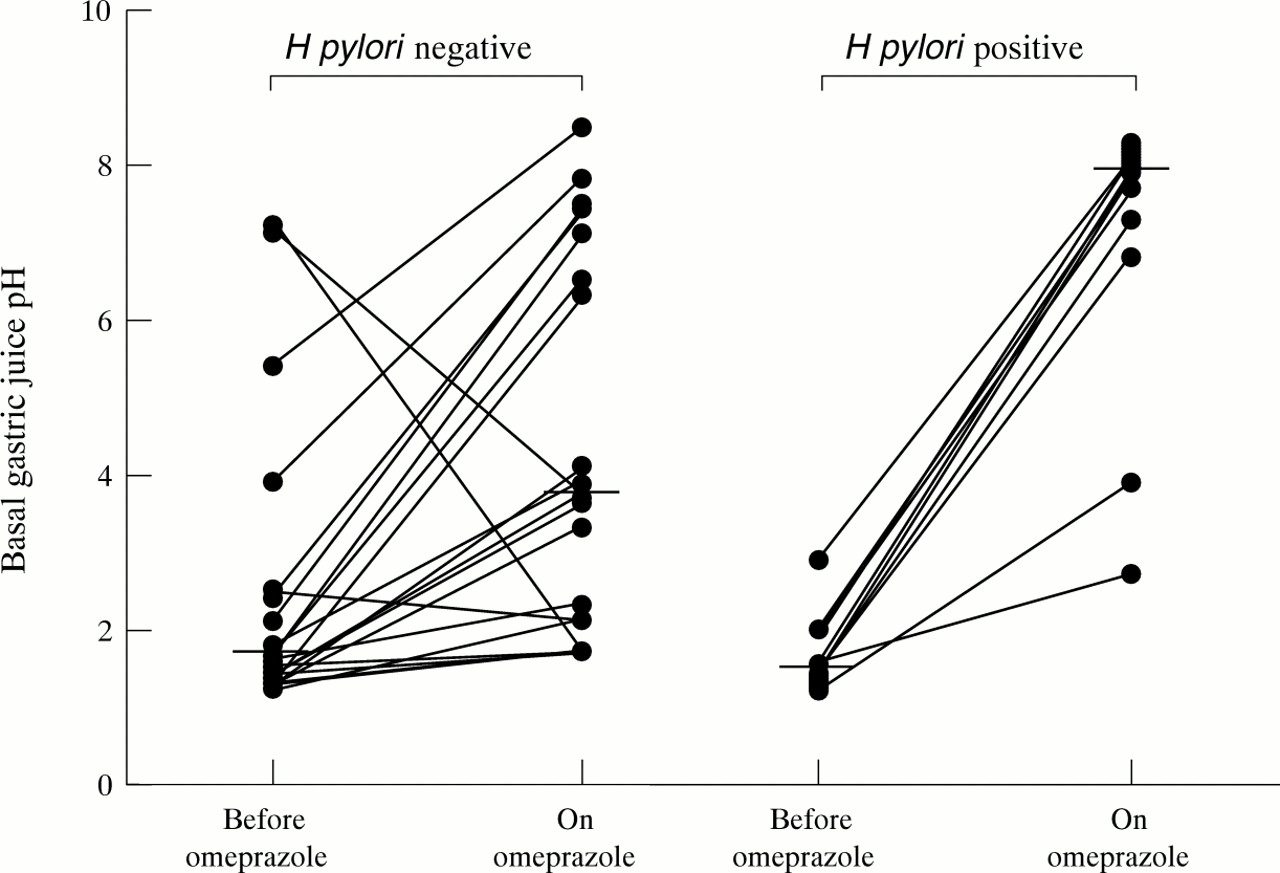
Pre-omeprazole, the median basal pH in the H pylori negative subjects was 1.6 (range 1.2–7.2), and that in the H pylori positive subjects was 1.6 (1.2–2.9) (p<0.84; fig 1). During omeprazole, the median basal pH in the H pylori negative subjects was 3.75 (1.7–8.5), and that in the H pyloripositive subjects was 7.95 (2.7–8.3) (p<0.002; fig1).

Basal fasting gastric juice pH in H pylori negative and positive subjects before and during omeprazole treatment. Medians are represented by horizontal bars.
BASAL PLASMA GASTRIN CONCENTRATIONS
Before omeprazole, the median basal gastrin in theH pylori negative subjects was 15 ng/l (range 5–90), which was not significantly different from that in theH pylori positive subjects (20 (5–75) ng/l; p<0.29; fig 2). Basal plasma gastrin was significantly higher on omeprazole than pre-omeprazole in both the H pylori negative subjects (p<0.001) and theH pylori positive subjects (p<0.003). However, during omeprazole, the median basal plasma gastrin concentration in the H pylori negative subjects (35 (5–120) ng/l), was considerably lower than that of theH pylori positive subjects (95 (30–400) ng/l; p<0.006; fig 2).

Basal plasma gastrin concentrations in the H pylori negative and positive subjects before and during omeprazole treatment. Medians are represented by horizontal bars.
BASAL ACID OUTPUT
Before omeprazole, the median basal acid output (BAO) in theH pylori negative subjects was 3.2 (0.0–9.7) mmol/h, which was the same as that of theH pylori positive subjects (3.2 (0.7–14.7) mmol/h; p<0.59; fig 3). BAO was lower on omeprazole than pre-omeprazole in both H pylori negative (p<0.001) and H pylori positive subjects (p<0.003). However, during omeprazole, the median BAO in theH pylori negative subjects was 0.3 (0.0–4.2) mmol/h, which was greater than that of theH pylori positive subjects (0.0 (0.0–1.2) mmol/h; p<0.009; fig 3).
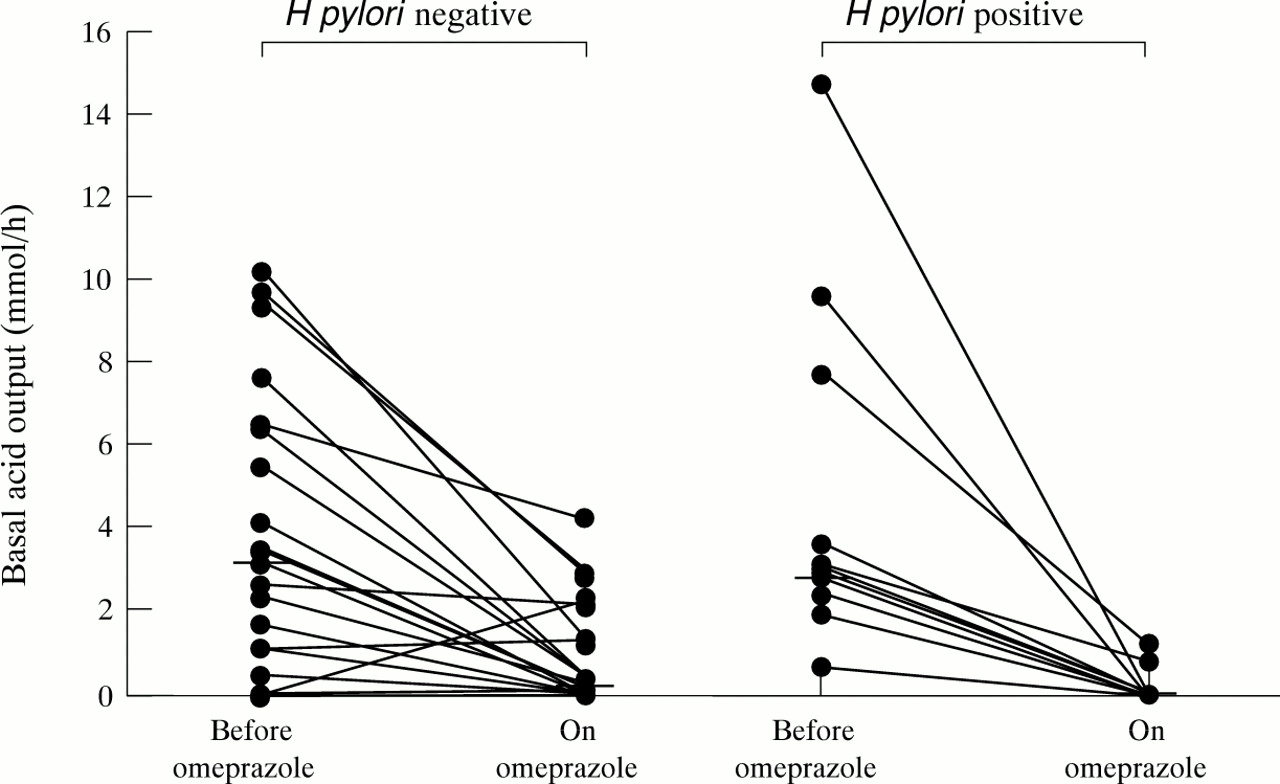
Basal acid output in H pylori negative and positive subjects before and during omeprazole treatment. Medians are represented by horizontal bars.
Before omeprazole, there was no difference in basal intragastric acidity between the two groups. However, on omeprazole, the median basal intragastric acidity of the H pylorinegative subjects (7.4 (0.0–36.4) mmol/l), was significantly greater than that of the H pylori positive subjects (0.0 (0.0–14.8) mmol/l; p<0.006).
During omeprazole, the median degree of inhibition of BAO in theH pylori negative subjects was 93.05% (−18.2% to 100%), which was less than that of theH pylori positive subjects (100% (75% to 100%); p<0.008). The median degree of inhibition of the basal volume of gastric juice secreted by the H pylorinegative subjects (43.85%) was also less than that of theH pylori positive subjects (61.8%), although this did not reach classical statistical significance (p<0.13). The median degree of omeprazole induced inhibition of basal intragastric acidity of the H pylorinegative subjects was 83.15% which was lower than that of theH pylori positive subjects (100%; p<0.007).
BASAL GASTRIC JUICE AMMONIUM CONCENTRATIONS AND AMMONIA OUTPUT
Before omeprazole, the median basal ammonium concentration in theH pylori negative subjects was 1023 (396–3210) μmol/l which was lower than that of theH pylori positive subjects (3285 (975–4590) μmol/l; p<0.002). During omeprazole, the median basal ammonium concentration of the H pylori negative subjects was 1088 (387–3465) μmol/l, which was also lower than that of the H pylori positive subjects (2220 (360–4035) μmol/l; p<0.003). This represents a difference in medians on omeprazole of 1.1 mmol/l.
From data for basal gastric juice volume and basal gastric juice ammonium concentration, the basal gastric juice ammonia output can be calculated, in a similar fashion to the calculation of gastric acid output, from the product of gastric juice volume and hydrogen ion concentration. Before omeprazole, the median basal ammonia output of the H pylori negative subjects (0.08 (0.01–0.59) mmol/h), was significantly lower than that of theH pylori positive subjects (0.28 (0.04–0.56) mmol/h; p<0.03). During omeprazole, the median basal ammonia output of the H pylori negative subjects (0.07 (0.01–0.36) mmol/h), was not significantly different from that of the H pylori positive subjects (0.13 (0.02–0.31) mmol/h; p<0.15).
MAXIMAL ACID OUTPUT
Before omeprazole, the median maximal acid output (MAO) of theH pylori negative subjects was 32.4 (17.9–53.0) mmol/h, which was similar to that of theH pylori positive subjects (32.2 (14.5–60.3) mmol/h; p<0.50; fig 4). During omeprazole, the median MAO of the H pylori negative subjects was 18.6 (3.2–39.0) mmol/h, which was greater than that of theH pylori positive subjects (6.0 (0.2–31.7) mmol/h; p<0.009; fig 4). MAO was lower on omeprazole than pre-omeprazole in both the H pylori negative (p<0.0009) and positive subjects (p<0.003).

Maximal acid output in the H pylori negative and positive subjects before and during omeprazole treatment. Medians are represented by horizontal bars.
Before omeprazole, there was no significant difference in intragastric acidity under maximal G-17 stimulation between the two groups. However, on omeprazole, the median intragastric acidity of theH pylori negative subjects (96.0 (39.2–119.6) mmol/l), was significantly greater than that of theH pylori positive subjects (43.2 (4.8–103.6) mmol/l; p<0.0008).
The median degree of omeprazole induced inhibition of MAO in theH pylori negative subjects (54.6% (−25.2% to 89.0%)) was less than that of the H pylori positive subjects (79.8% (27.3% to 99.4%); p<0.003). The median degree of omeprazole induced inhibition of the volume of gastric juice secreted in the H pylorinegative subjects (33.0% (−82.6% to 81.8%)) was less than that of the H pylori positive subjects (50.0% (1.3% to 84.4%); p<0.04). The median degree of omeprazole induced inhibition of intragastric acidity of the H pylori negative subjects (29.8% (−16.6% to 59.5%)) was less than that of the H pylori positive subjects (61.7% (26.3% to 96.1%)); (p<0.001).
GASTRIC JUICE AMMONIUM CONCENTRATION AND AMMONIA OUTPUT DURING MAXIMAL G-17 STIMULATION
Before omeprazole, the median ammonium concentration in theH pylori negative subjects was 661.5 (276–1425) μmol/l, which was lower than that in theH pylori positive subjects (1958 (178–5670) μmol/l; p<0.009). During omeprazole, the median ammonium concentration in the H pylori negative subjects was 825 (378–1485) μmol/l, which was also lower than that in the H pylori positive subjects (2025 (915–8055) μmol/l; p<0.0002). This represents a difference in medians on omeprazole of only 1.2 mmol/l.
Before omeprazole, the median ammonia output of theH pylori negative subjects (0.16 (0.07–0.53) mmol/h), was significantly lower than that of theH pylori positive subjects (0.51 (0.04–1.34) mmol/h; p<0.04). During omeprazole, the median ammonia output of the H pylori negative subjects (0.16 (0.07–0.48) mmol/h) was not significantly different from that of the H pylori positive subjects (0.23 (0.10–1.07) mmol/h; p<0.16).
SUBMAXIMAL ACID OUTPUTS DURING G-17 STIMULATION
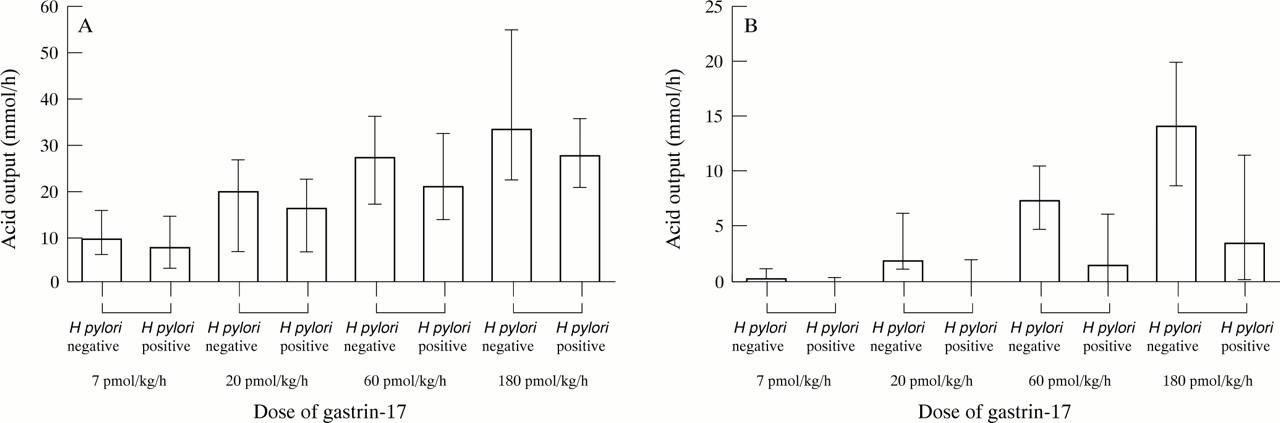
Table 1 shows median acid outputs at infusion rates of 7, 20, 60, and 180 pmol/k/h of G-17. Before omeprazole there were no significant differences at any G-17 infusion rate. However, on omeprazole, the acid outputs of the H pylori negative subjects were significantly greater at all infusion rates (fig5).
Acid output at submaximal doses of gastrin 17 in H pylori negative and positive subjects before and during omeprazole

Median acid outputs (and ranges) to the submaximal doses of G-17 in H pylori negative and positive subjects (A) before and (B) during omeprazole treatment.
Table 2 shows the median intragastric acidities in both groups, at each of the submaximal doses of G-17. The H pylori negative subjects had a significantly lower median degree of omeprazole induced inhibition of acid secretion than theH pylori positive subjects at each of the submaximal doses of G-17: 96.45% versus 100% at 7 pmol/kg/h (p<0.05); 88.6% versus 99.5% at 20 pmol/kg/h (p<0.02); 74.05% versus 93.6% at 60 pmol/kg/h (p<0.009); and 56.15% versus 86.95% at 180 pmol/kg/h (p<0.01).
Intragastric acidity at submaximal doses of gastrin 17 in H pylori negative and positive subjects before and during omeprazole
Before omeprazole, there were trends to a significantly higher plasma gastrin in the H pylori positive subjects at the submaximal G-17 doses of 7 and 20 pmol/kg/h and no significant differences at 60 and 180 pmol/kg/h (table 3). However, during omeprazole, the H pylori positive subjects had a significantly higher plasma gastrin at 7 and 20 pmol/kg/h, a trend to a difference at 60 pmol/kg/h, but no significant difference at 180 pmol/kg/h (table 3). Due to the higher gastrin concentrations in the H pylori positive subjects during the lower G-17 infusion doses, the acid response was plotted against gastrin concentration (fig 6). This showed that on omeprazole, theH pylori positive subjects had a notably reduced acid response across the full range of gastrin concentrations when compared with the H pylori negative subjects.
Serum gastrin concentrations during infusions of gastrin 17 in H pylori negative and positive subjects before and during omeprazole

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Median plasma gastrin concentration versus median acid output curves for the H pylori negative and positive subjects before and during omeprazole treatment.
Discussion
Previous studies have shown that during omeprazole treatment, intragastric pH is more notably elevated in H pylori positive versus negative healthy subjects.21In addition, intragastric pH on omeprazole is higher inH pylori infected subjects than in the same subjects after the infection has been eradicated.22 ,23 In these studies, the mean 24 hour pH in H pylori infected subjects on omeprazole was 5.0–5.5, compared with 3.0–3.5 in H pylori negative or eradicated subjects.21-23 This difference in pH represents a very small difference in hydrogen ion concentration of less than 1 mmol/l. This has led the groups which have documented the pH phenomenon to conclude that it may represent nothing more than neutralisation of intragastric acid by H pylori produced ammonia.22 ,23 Our current studies investigated whether H pylori status might be affecting the degree of suppression of gastric acid secretion produced by omeprazole, which would make the phenomenon of greater clinical significance.
Our study confirms these previous pH observations. The median fasting pH in the H pylori negative subjects on omeprazole was 3.75 versus 7.95 in the H pylori positive subjects. Only 25.0% of theH pylori negative subjects had neutral basal pH values 24 hours after the previous dose of the PPI, compared with 83.3% of the infected subjects. The previous studies reporting the influence of H pylori status on the pH raising efficacy of omeprazole had been conducted after seven days of dosing.21-23 Our present study was performed after at least six weeks of omeprazole and indicates that the phenomenon persists with longer courses of therapy.
In addition to measuring fasting intragastric pH, we performed detailed studies of basal, submaximal, and maximal acid output. Prior to commencing omeprazole, there were no differences between theH pylori positive and H pylori negative healthy subjects with respect to basal and G-17 stimulated maximal acid output. This is consistent with our previous studies showing that increased acid secretion induced byH pylori infection is mainly confined to duodenal ulcer patients.30 Submaximal G-17 stimulated acid output was slightly lower in the H pyloripositive than in the H pylori negative subjects. This was more apparent when the acid output was assessed against the gastrin concentration than against the G-17 dose, as the latter does not take into account the higher endogenous gastrin level in the H pylori positive subjects. The reduced acid response to gastrin in the infected subjects is consistent with our recent report of reduced sensitivity to G-17 inH pylori infected healthy volunteers.30
Marked differences in acid secretion were apparent between theH pylori positive and negative subjects on omeprazole. The median BAO in the H pyloripositive subjects was 0.0 mmol/h, compared with 0.3 mmol/h in the uninfected subjects. This difference in BAO could not be explained by any neutralising effect of ammonia produced by H pylori. The median ammonia output on omeprazole was 0.13 mmol/h in the H pylori positive and 0.07 mmol/h in the H pylori negative subjects. This represents a difference in ammonia output of only 0.06 mmol/h, which is fivefold less than the difference in BAO. Furthermore, the greater degree of inhibition of BAO in the H pyloripositive versus negative subjects on omeprazole was due to a greater degree of inhibition of both volume of gastric juice secreted and its acidity. This provides further evidence that the difference in acid measured was due to greater inhibition of acid secretion in theH pylori positive subjects and not merely neutralisation by either ammonia or other neutralising factors.
MAO to G-17 was also considerably lower in the H pylori positive versus negative subjects on omeprazole, being 6.0 mmol/h and 18.6 mmol/h respectively. As with BAO, this difference in MAO of 12.6 mmol/h could not be explained by a neutralising effect of ammonia produced by H pylori. The median ammonia output during maximal G-17 stimulation of omeprazole was 0.23 mmol/h in the H pylori positive subjects and 0.16 mmol/h in the uninfected subjects. This difference of 0.07 mmol/h between the infected and uninfected subjects could not explain the more than 180-fold greater 12.6 mmol/h difference in median MAO. Furthermore, this difference in acid output on omeprazole was also due to a greater degree of inhibition of volume, as well as acidity, and this again excludes neutralisation by ammonia, or indeed other neutralising substances, as a feasible explanation of this observation.
Acid output on omeprazole in response to submaximal stimulation with G-17 showed a very notable difference between the H pylori positive and negative subjects (figs 5 and 6). At a dose of 60 pmol/kg/h, the median acid output in the H pylori negative subjects was 7.4 mmol/h compared with 1.6 mmol/h in the H pylori positive subjects. At a dose of 180 pmol/kg/h, the corresponding values were 14.2 and 3.6 mmol/h respectively. Significant differences in acid output were also apparent at 7 and 20 pmol/kg/h. Furthermore, at 7 and 20 pmol/kg/h of G-17 during omeprazole, the plasma gastrin concentrations achieved during the gastrin infusion were significantly higher in theH pylori positive versus theH pylori negative subjects, to some degree masking the true magnitude of the difference of the acid response to gastrin at these doses of G-17. This difference in gastrin concentrations between the H pylori negative and positive subjects during the lower doses of the G-17 infusion can be explained by the contribution of the higher endogenous gastrin levels in the infected subjects.
Our studies have thus shown that omeprazole produces more notable suppression of BAO, submaximal acid output, and MAO inH pylori positive than inH pylori negative subjects. The degree of inhibition of BAO was 100% in the H pyloripositive versus 93.35% in the negative subjects, 93.6% versus 74.05% for submaximal (60 pmol/kg/h) acid output, and 79.8% versus 54.6% respectively for MAO.
Sensitivity to gastrin stimulation can be assessed by plotting plasma gastrin concentration against acid output at the various doses of G-17 and calculating the concentration of G-17 to produce half maximal response.30 However, in this present study, the degree of acid suppression, particularly in the H pylori positive subjects on omeprazole, made it impossible to produce a concentration/acid response curve of sufficient accuracy to calculate the sensitivity to gastrin. Despite this, the observation that the gastrin concentration/acid response curve in theH pylori positive subjects on omeprazole is shifted notably to the right, compared with that of theH pylori negative subjects on omeprazole, is consistent with the former having a lower sensitivity to gastrin on omeprazole.
All of the previously published studies of the influence ofH pylori status on the response to omeprazole only measured intragastric pH. These studies concluded that the difference in pH could be largely explained by neutralisation of intragastric acid by H pylori produced ammonia. Our current study indicates that there is a notable difference in the degree of suppression of gastric acid secretion inH pylori positive versus negative subjects and that neutralisation by ammonia production cannot explain more than 20% of the difference in BAO or 0.6% of the difference in MAO. Similarly, the fact that the volume of gastric juice secreted is affected, as well as its acidity, indicates that the presence of any other neutralising substances, such as enhanced mucosal bicarbonate production31 or H pylorirelated duodenogastric reflux32 cannot explain the observation.
The previously reported studies were performed after seven days of therapy, whereas our present study examined subjects after six to eight weeks of treatment, being representative of a typical course in clinical practice. It is possible that intragastric ammonia levels and degree of inhibition of acid varies slightly with different duration of treatment.
Our findings thus indicate that some interaction is occurring betweenH pylori infection and omeprazole treatment which is potentiating the antisecretory efficacy of the drug. A plausible explanation for this is the intense inflammation of the oxyntic mucosa which develops in H pyloripositive subjects during PPI treatment.18-20 This increased gastritis is likely to impair the function of the oxyntic mucosa and thereby supplement the pharmacological effect of the drug. Recent observations by ourselves and others33-35 that there is a negative correlation between H pylori associated inflammation of the oxyntic mucosa and pentagastrin stimulated peak acid output in H pylori infected patients is consistent with this theory. Furthermore, eradication of the organism and the accompanying resolution of oxyntic inflammation result in prompt recovery of acid secretory function.36 H pyloriinduced inflammation results in an increased production of a variety of cytokines, including interleukin 1 (IL-1), which has been shown to a very powerful inhibitor of acid secretion.37-39 An alternative or additional explanation for the more notable inhibition of acid secretion in H pylori positive subjects is that ammonia produced by the urease inH pylori is able to penetrate the oxyntic mucosa during omeprazole treatment due to more being in the more lipophilic unionised form at the higher intragastric pH. This could allow increased delivery of NH4 + ions close to the proton pumps, where they can act as K+surrogates,40 and lead to uncoupling of the proton pumps.41 We believe that the more profound inhibition of acid secretion in H pylori positive subjects on PPI treatment is unlikely to be due to acid inhibitory products of the bacterium,42-44 as such treatment does not increase the density of bacterial colonisation of the oxyntic mucosa.20 However, one cannot exclude such products being able to gain greater access to the acid secreting cells when acid secretion is inhibited or their being more active at less acidic pH.
Our observation that the influence of H pyloristatus on the pH elevating effect of omeprazole is due to a difference in the actual acidity and volume of the gastric secretion increases the clinical importance of the phenomenon. Gastric acid is an important element of the phylogenetically conserved non-specific immune system.45 ,46 The H pyloripositive patients rendered profoundly hypochlorhydic by PPIs are therefore likely to be at increased risk of enteric infections, as susceptibility to such infection is known to exist in other low acid states.47-52 Certainly, increased susceptibility to enteric infection on omeprazole has been reported,53 but the H pylori status of the patients was not known. Our own group has recently reported a greater number of non-H pylori bacteria colonising the gastric juice of H pylori positive versus negative subjects during omeprazole treatment.54 Such bacterial colonisation may also result in the intragastric synthesis of potentially carcinogenic nitrosoamines.55
The findings from our present study that the degree of inhibition of both the volume and acidity of gastric secretion by omeprazole is considerably less in the H pylori negative than positive subjects makes it highly likely that its efficacy in controlling acid/peptic disease will also be less inH pylori negative subjects. All the clinical studies to date which have assessed the antisecretory efficacy of PPI treatment have involved groups which have been either predominantly (ulcer patients) or partially H pyloripositive.6-8 56-63 The current literature on the antisecretory efficacy of PPI treatment may thus overestimate its efficacy in the H pylori negative population. There have been recent reports of difficulty in controlling intragastric acidity in some GORD subjects with PPI treatment.64 This may be due to reduced efficacy inH pylori negative subjects. Whether increasing the dose of the PPI will achieve increased control is at present unclear and will need to be addressed in H pylori negative subjects.
In summary, H pylori status has a major influence on the inhibition of acid secretion produced by PPI treatment. A more profound inhibition of acid secretion is seen inH pylori positive subjects and this cannot be explained by acid neutralisation, either by ammonia or any other substances. While this increased antisecretory effect will facilitate the control of acid/peptic disease, it will also predispose to enteric infections and gastric colonisation by nitrosating bacterial species.
Acknowledgments
We gratefully acknowledge the kindness of the Scottish National Blood Transfusion Service in their provision of serum albumin. We are also most grateful to Dr Andrew Kelman for his kind advice on appropriate pharmacokinetic modelling.
Abbreviations
- BAO
- basal acid output
- ECL
- enterochromaffin-like
- G-17
- gastrin 17
- GORD
- gastro-oesophageal reflux disease
- MAO
- maximal acid output
- PPI
- proton pump inhibitor