Article Text

Abstract
BACKGROUND Neutrophil-endothelial cell interactions are thought to play a critical role in the pathophysiology of non-steroidal anti-inflammatory drug (NSAID) induced gastropathy.
AIMS To optimise a mouse model of NSAID induced gastropathy and to evaluate the importance of adhesion molecules using adhesion molecule deficient mice.
METHODS Gastropathy was induced in C57BL/6 mice or their adhesion molecule deficient counterparts via oral administration of indomethacin (20 mg/kg). Lesion scores, mucosal permeability, and histopathology were used to assess gastric mucosal injury.
RESULTS Intragastric administration of indomethacin induced linear haemorrhagic mucosal lesions, primarily in the corpus of the stomach that were first observed at six hours. These lesions continued to develop over the next six hours with maximal lesion scores and mucosal permeabilities at 12 hours. When indomethacin was administered to mice deficient in CD18, intercellular adhesion molecule 1 (ICAM-1), or P-selectin, there were significant decreases in lesion scores compared with their C57BL/6 controls. In addition, mucosal permeabilities were found to be significantly lower in CD18 or ICAM-1 deficient mice observed at 12 hours.
CONCLUSION Certain leucocyte and endothelial cell adhesion molecules are important determinants for full expression of indomethacin induced gastropathy. It is proposed that this modification of the mouse model may be useful for the investigation of other pathophysiological mechanisms of NSAID induced gastropathy.
- indomethacin
- gastropathy
- cyclooxygenase
- intercellular adhesion molecule
- VCAM
- vascular cell adhesion molecule
- P-selectin
Abbreviations used in this paper
- NSAID
- non-steroidal anti-inflammatory drug
- COX
- cyclooxygenase
- ICAM
- intercellular adhesion molecule
- PMN
- polymorphonuclear leucocyte
- VCAM
- vascular cell adhesion molecule
- TNF
- tumour necrosis factor
Statistics from Altmetric.com
- indomethacin
- gastropathy
- cyclooxygenase
- intercellular adhesion molecule
- VCAM
- vascular cell adhesion molecule
- P-selectin
A major limitation to the use of non-steroidal anti-inflammatory drugs (NSAIDs) for the treatment of chronic inflammatory diseases (for example, rheumatoid arthritis) is the development of gastric mucosal injury. Previous studies have suggested that NSAIDs induce this gastropathy by inhibiting cyclooxygenase (COX) mediated production of “protective” prostaglandins.1 ,2 However, several recent studies suggest that the mechanism by which NSAIDs promote gastric injury may be much more complex than originally thought. For example, Ligumskyet al have shown that rectal administration of certain NSAIDs inhibited prostaglandin production by greater than 95% yet did not induce gastric ulcers.3 In addition, Langenbach et al have shown that deletion of the Ptgs 1 gene encoding for the COX-1 isoenzyme in mice did not result in the development of spontaneous gastric ulcers.4 Indeed, these COX-1 deficient animals seemed to be less sensitive to NSAID induced gastropathy than their age matched controls.4
It is becoming increasingly appreciated that neutrophilic polymorphonuclear leucocytes (PMNs) may play an important role in the pathogenesis of NSAID induced gastropathy. Wallaceet al have shown that NSAID induced gastric mucosal injuries are significantly reduced by immunoneutralisation of CD18, intercellular adhesion molecule 1 (ICAM-1), P-selectin, and to a lesser extent E-selectin, or by rendering animals neutropenic via the intravenous administration of anti-PMN antiserum.5-7We have recently shown, using a newly developed dual radiolabelled monoclonal antibody technique for quantification of surface expression of endothelial cell adhesion molecules in vivo, significant increases of gastric mucosal surface expression of P-selectin and ICAM-1 in an established rat model of NSAID induced gastropathy.8
A number of questions remain regarding the mechanisms by which NSAID induced leucocyte–endothelial cell interactions in the gastric mucosal microcirculation lead to epithelial cell necrosis and ulcer formation. The recent development of several different strains of genetically engineered mice with specific gene deletions (knock outs) or overexpressions (transgenic) makes it theoretically possible to identify more accurately specific adhesion molecules or inflammatory mediators (cytokines, nitric oxide, arachidonate metabolites, etc) that may be involved in NSAID induced gastropathy.9-11 Unfortunately, only a few studies have used a mouse model of gastropathy in which NSAIDs were administered orally. Indeed, the few studies that have been reported using doses of NSAIDs comparable to those used in rat studies showed only modest gastric injury.4 ,12 ,13 Therefore, we wished to optimise an existing mouse model of gastropathy using oral doses of NSAIDs comparable to those used in the rat study (20 mg/kg)5 ,7 ,8 and evaluate the importance of certain adhesion molecules in this model using genetically engineered adhesion molecule deficient mice.
Methods
INDOMETHACIN INDUCED GASTROPATHY
CD18, ICAM-1, and P-selectin deficient mice bred on a C57BL/6 background were obtained from Pharmacia-Upjohn Laboratories (Kalamazoo, Michigan, USA).9-11 The mutants were all developed in the 129 mouse strain and the number of backcrosses to C57BL/6 ranged between eight (P-selectin) and 10 (CD18 and ICAM-1) generations. Immunohistochemistry and flow cytometry have been used to verify the respective adhesion molecule deficiency, and functional studies have shown a notably attenuated recruitment of adherent (CD18 and ICAM-1) or rolling (P-selectin) leucocytes in these mutants.14 Wild type C57BL/6 mice were purchased from Harlan Laboratories (Frederick, Maryland, USA). These animals were housed under specific pathogen free conditions. Different doses of indomethacin (5, 10, 20, and 40 mg/kg, dissolved in 0.25 ml of 5% sodium bicarbonate) or vehicle (0.25 ml of 5% sodium bicarbonate) were administered orally to wild type or adhesion molecule deficient mice (25–28 g, 12–15 weeks of age) following the deprivation of food, but not water, for 10–16 hours. Measurement of mucosal permeabilities and lesion scores were performed 0–18 hours post indomethacin administration. All procedures involving the use of animals were approved by, and in accordance with the guidelines of the Louisiana State University Medical Centre Animal Care and Use Resources Committee.
MEASUREMENT OF MUCOSAL PERMEABILITY
Prior to and at 3, 6, 12, and 18 hours following indomethacin administration, mice were anaesthetised via an intramuscular injection of ketamine and xylazine at a dose of 150 and 7.5 mg/kg, respectively. Body temperature was maintained at 37°C with a thermistor controlled water pad (Aquamatic K-Modules K-20; Baxter, Valencia, California, USA). The right carotid artery and left jugular vein were cannulated. A laparotomy was performed using a midline abdominal incision. The stomach was cannulated at the abdominal oesophagus using polyethylene tubing (Becton Dickinson and Company, Sparks, Maryland, USA; inner diameter 0.58 mm) for continuous infusion (0.4 ml/min) of warm (37°C) saline (pH 3.5). The stomach was also cannulated at the proximal portion of the duodenum into the proximal region of the gastric pylorus using polyethylene tubing (inner diameter 1.67 mm) for collecting the solution. The perfused stomach and abdominal contents were kept moist with saline soaked gauze and covered with a clear plastic sheet to minimise evaporation and tissue dehydration. The luminal content of the stomach was removed by preperfusion of warm saline (pH 3.5) for 15 minutes.
Gastric mucosal permeability was determined using the blood to lumen clearance of 51Cr labelled EDTA as modified for mice from the conventional method as described previously.15Following 25 μCi of 51Cr-EDTA (DuPont de Nemours & Co. Inc., Birmington, Delaware, USA) injection, 0.2 μCi/min of51Cr-EDTA was continuously infused via the jugular vein catheter. Following a 25 minute equilibration period, the perfusate was collected every 10 minutes for 30 minutes for the appearance of51Cr-EDTA. Plasma samples were obtained at 0 and 30 minutes for use as reference counts. Radioactivity in each sample was determined using a multichannel gammacounter (Wallac 1282 Compugamma). Blood to lumen clearance of 51Cr-EDTA was calculated using the following formula: Clearance (μl/min/g wt) = (Cper × Q)/(Cpl × W) × 1000 where Cper and Cpl represent counts per minute per ml of 51Cr-EDTA in the lumen perfusate and plasma, respectively; Q represents the luminal perfusion rate (400 μl/min); and W represents the weight of the stomach. Mucosal permeability was determined from the mean of the four clearance values.
LESION SCORING, TISSUE PREPARATION, AND BIOCHEMICAL ANALYSIS
Following measurement of mucosal permeability, the animals were killed with an overdose of pentobarbital sodium (Butler, Columbus, Ohio, USA), and the perfused stomachs were excised. The stomachs were opened along the greater curvature and examined. As indomethacin produces linear ulcers, lesion scores of each animal were expressed as the sum of the length of lesions as described previously.8
Each stomach was then sectioned, fixed, dehydrated, and embedded in JB-4 (Polysciences Inc., Warrington, Pennsylvania, USA). Sections (2.5 mm) were cut with glass knives, stained with haematoxylin and eosin, and evaluated for histopathology.
STATISTICS
All values are presented as means (SEM). The data were analysed using the one way analysis of variance followed by the Student-Newman-Keuls multiple comparisons test. Statistical significance was set at p<0.05.
Results
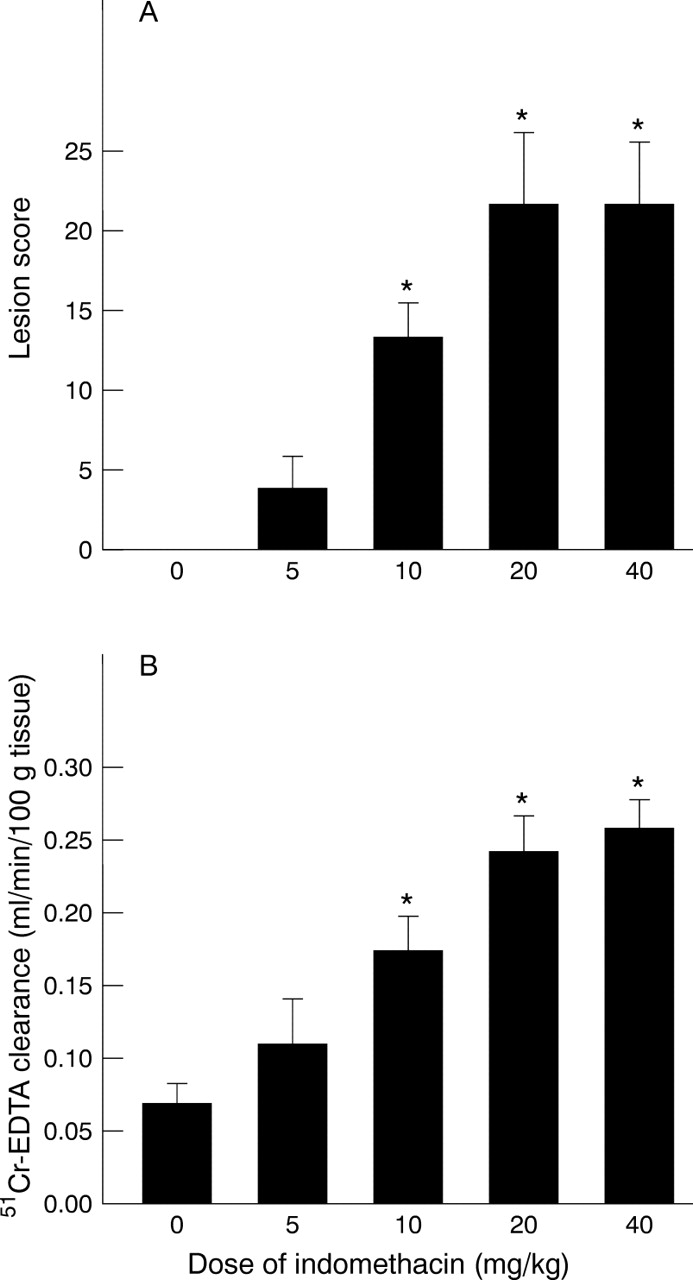
In a first series of studies, intragastric administration of indomethacin (20 mg/kg) induced linear haemorrhagic lesions, primarily in the corpus of the stomach, that were first observed macroscopically at six hours after administration. These haemorrhagic erosions continued to develop over the next six hours and were characterised histologically by mucosal injury (oedema, necrosis, and exfoliation of the mucosal epithelial cells into the gastric lumen), haemorrhage, and formation of a “mucoid cap” (a layer of mucus, fibrin, and necrotic tissue) in a manner similar to the lesions induced by indomethacin in the rat (fig 1A, B). There was no histological evidence of significant neutrophil infiltration into the gastric mucosal interstitium. Lesion score increased at six hours following indomethacin administration and continued to increase such that maximal lesion scores were also observed at 12 hours post indomethacin treatment (fig 2A). Mucosal permeabilities increased in a similar manner, peaking at 12 hours post indomethacin administration (fig 2B). Lesion scores and mucosal permeabilities increased in a dose dependent manner using indomethacin doses ranging from 5 mg/kg to 40 mg/kg (fig 3A, B). As values for mucosal lesions and permeabilities were most consistent at a dose of 20 mg/kg and a time of 12 hours, this dose and time were used for all additional studies.

Histopathology of C57BL/6 mouse gastric mucosa resulting from indomethacin treatment. (A) Normal (untreated) mucosa. Typical distribution of apical located surface and mucous neck cells and more basal located chief and parietal cells. (B) Indomethacin treated mucosa of the wild type C57BL/6 mouse. Typical lesion showing erosion of the mucosa with predominant loss of the surface and mucous neck cells and loss of the integrity of the apical region of the glands. (C) Indomethacin treated mucosa of the CD18 deficient C57BL/6 mouse. Typical mucosa showing substantial protection to indomethacin treatment. The apical region of the glands appears disorganised, but does not exhibit erosion. (D) Indomethacin treated mucosa of the P-selectin deficient C57BL/6 mouse. Typical mucosa showing moderate protection to indomethacin treatment. The apical region of the glands appears disorganised with some general loss of surface mucous and mucous neck cells. Distinct regions of erosive lesion are not apparent. (E) Indomethacin treated mucosa of the ICAM-1 deficient C57BL/6 mouse. The absence of ICAM-1 expression had little protective effect. Typical lesions are found showing substantial erosion of the mucosa with loss of the integrity of the glands and loss of cells. However, ICAM-1 deficiency seemed to reduce the number of lesions.

Time course of gastric mucosal lesion score (A) and mucosal permeability (B) following oral administration of indomethacin (20 mg/kg) to healthy wild type (C57BL/6) mice. *p<0.05 versus 0 hour.

Gastric mucosal lesions (A) and mucosal permeability (B) induced by varying doses of indomethacin to healthy wild type mice. *p<0.05 versus control.
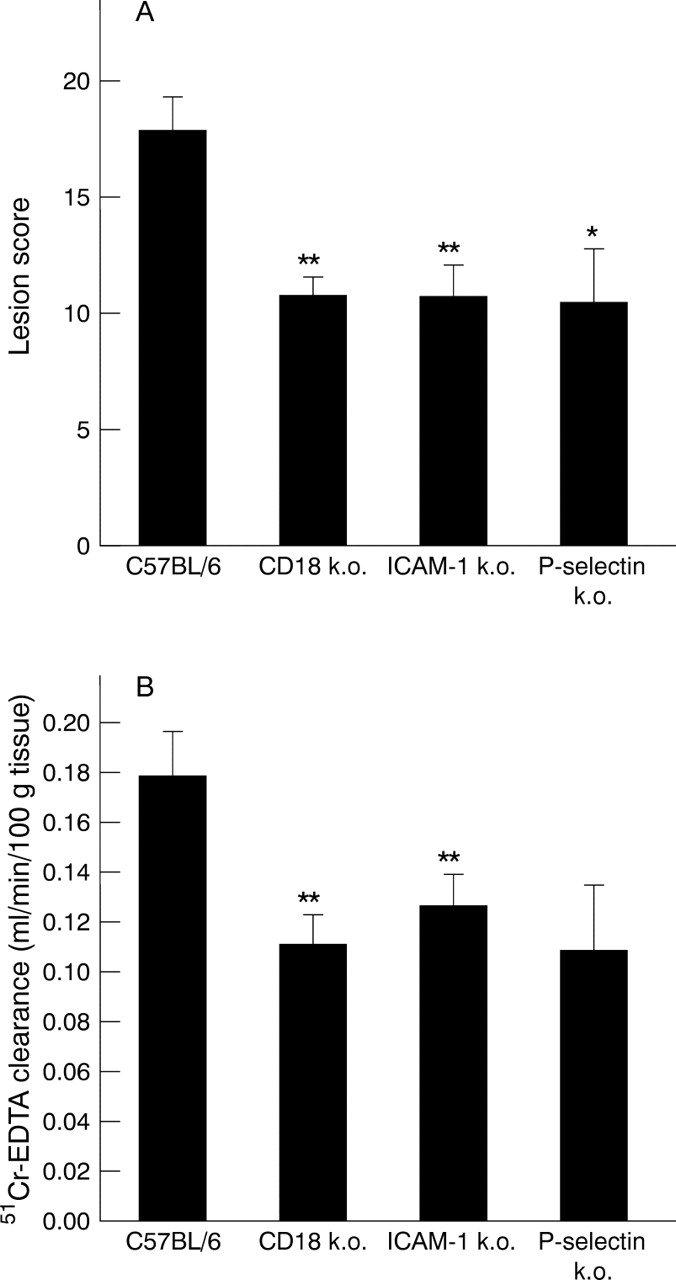
In a second series of studies, intragastric administration of indomethacin (20 mg/kg) to mice deficient in CD18 (fig 1C), ICAM-1 (fig1E), or P-selectin (fig 1D) resulted in improvement in mucosal histopathology. ICAM-1 deficient mice seemed to have fewer numbers of lesions, whereas the P-selectin and CD18 deficient response to indomethacin was less severe erosions and/or ulcerations. Furthermore, animals deficient in the three adhesion molecules also exhibited significant reductions in lesion scores of approximately 40% compared with their C57BL/6 controls (fig 4A). In addition, gastric mucosal permeabilities were significantly reduced in CD18 or ICAM-1 deficient mice by 38% and 29% at 12 hours post indomethacin administration respectively (fig 4B). Gastric mucosal permeabilities were also reduced in P-selectin deficient mice at 12 hours post indomethacin administration, but these changes were not significant (p=0.06).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Gastric mucosal lesion score (A) and mucosal permeability (B) in wild type (C57BL/6), ICAM-1, CD18, and P-selectin deficient mice 12 hours following oral administration of indomethacin (20 mg/kg). *p<0.05 and **p<0.01 versus C57BL/6 mice.
Discussion
Recent studies from our laboratory,8 showing significant increases of gastric mucosal surface expression of P-selectin and ICAM-1 in an established rat model of NSAID induced gastropathy, coupled to previous work by Wallace and colleagues5-7 strongly suggest that PMN-endothelial cell interactions play a critical role in the pathophysiology of NSAID induced gastropathy. Although much has been learned from these studies, the mediators and mechanisms responsible for NSAID induced gastric mucosal injury remain to be defined. Unfortunately many of the immunological and molecular probes that may be used to assess the role of different cytokines, mediators (arachidonate metabolites, nitric oxide), and adhesion molecules are not available for studies in the rat. Furthermore, the recent development of several different genetically engineered mice makes this species particularly attractive as an animal model to define the pathogenetic mechanisms responsible for NSAID induced gastropathy. In the present study, we have modified and optimised an existing mouse model of NSAID induced gastropathy and evaluated the relative importance of specific adhesion molecules in this model, using mice deficient in three different adhesion molecules.9-11
We found that orally administered indomethacin induced macroscopic and histological gastric mucosal lesions similar to those induced in a well established rat model of gastropathy.8Indomethacin produced haemorrhagic linear lesions primarily in the corpus region which were limited to the mucosal layer (fig 1). Although similar doses of indomethacin were used in the present study, a major difference between the mouse and rat models of gastropathy is the time to develop the macroscopic and histological lesions.4 ,12 ,13 We found that the macroscopic lesions began to develop at approximately six hours following intragastric administration of indomethacin in the mouse model versus one to two hours in the rat model.8 Mucosal lesions continued to develop over the next six hours in the mouse, resulting in maximal macroscopic and histological injury at 12 hours following indomethacin administration. This indomethacin induced mucosal injury seemed to continue for an additional 12 hours resulting in active gastritis at 24 hours post NSAID administration (data not shown). Studies from our laboratory, as well as others, show that the same dose of indomethacin (20 mg/kg) induces maximal gastric mucosal injury at three to six hours in rats.8 The reasons for these rather significant differences between mouse and rat are not known but may be due to differences in gastric mucosal defence systems (gastric mucosal blood flow, mucus secretion, etc) or to differences in absorption and/or metabolism of indomethacin. For example, if the ulcerogenic property of indomethacin is dependent on the maintenance of a given concentration for a period of time, and the rate of destruction or excretion of the drug is proportional to the metabolic rate of the particular species in question, then mice would be expected to metabolise indomethacin two times more rapidly than rats because of their higher metabolic rate.16
In order to determine whether leucocyte-endothelial cell interactions play a role in the pathogenesis of gastric ulceration in our murine model, we quantified the extent of gastric mucosal injury in CD18, ICAM-1, or P-selectin deficient mice and compared these data to those obtained with C57BL/6 controls. Previous studies have shown that these same CD18, ICAM-1, and P-selectin deficient mice exhibit a blunted response to leucosequestration in response to ischaemia and reperfusion induced injury in vivo.14 Interestingly, we found that adhesion molecule deficient mice exhibited a 40% inhibition in indomethacin induced mucosal injury compared with the 80–90% reduction in injury using anti-ECAM monoclonal antibodies as previously reported in the rat model.7 ,8 CD18 and P-selectin deficiency seem to suppress the development of erosive lesions; however, ICAM-1 deficiency seems to protect by reducing the number or extent of lesions (fig 1). There are several possibilities which may explain these differences. For example, it has been shown that in certain adhesion molecule deficient mice, other adhesion molecules (for example, P-selectin in CD18 deficient mice) are known to be enhanced, suggesting some sort of compensatory response.17 Another possibility relates to the longer time required for the mouse gastric mucosa to become injured in response to intragastric indomethacin. It may be that other adhesion molecules such as VCAM-1 and E-selectin are more important in the pathophysiology of mouse gastric ulceration. Finally, differences in the synthesis of other factors such as tumour necrosis factor α (TNF-α) may be altered in the gene deleted mice compared with their wild type controls.18
Another interesting characteristic of this model (as well as the rat model) is the lack of significant infiltration of PMNs or other leucocytes into the gastric mucosal interstitium in response to NSAIDs.8 These data suggest that the mechanisms by which adhesion of PMNs to the post-capillary venules induces epithelial cell necrosis and ulcer formation are much more complicated than originally thought. There are several possible mechanisms by which NSAID induced PMN adhesion may contribute to the pathogenesis of gastric mucosal injury. Firstly, the factors that trigger the adhesion of PMNs to the gastric microvasculature (for example, TNF-α, leukotriene B4) are likely to trigger the metabolic activation of these cells, leading to the liberation of potentially injurious and diffusible agents such as reactive oxygen metabolites (for example, H2O2, HOCl) and/or proteases. These substances may mediate some of the endothelial and epithelial injury caused by NSAIDs. Indeed, there is good evidence that reactive oxygen metabolites contribute to the mucosal injury observed after NSAID administration to rats.19 A second mechanism may involve PMN-endothelial adhesion as well as leucocyte and/or platelet aggregation, leading to obstruction of capillaries and a consequent reduction in gastric mucosal blood flow. This PMN dependent ischaemic event could ultimately promote mucosal injury. Indeed, it has been reported that NSAIDs reduce gastric mucosal blood flow which seems to occur subsequent to the formation of white thrombi in the gastric microcirculation.20 It should be noted however, that we have been unable to observe a significant reduction in rat gastric mucosal blood flow at any time following intragastric administration of indomethacin.8 Finally, with the recent realisation that cell adhesion molecules also transmit signals from outside the cell to inside,21-23 one could argue that engagement of certain adhesion molecules with their counter receptors may promote one or more signalling pathways that ultimately lead to mucosal injury.
Taken together, we propose that CD18, ICAM-1, and P-selectin play important roles in NSAID induced gastropathy in this mouse model of gastropathy. However, the mechanism by which adhesion of PMNs to microvascular endothelial cells induces gastric mucosal injury has still to be defined.
Acknowledgments
This work was supported in part by a grant from the National Institutes of Health DK43785 (Project 6).
Abbreviations used in this paper
- NSAID
- non-steroidal anti-inflammatory drug
- COX
- cyclooxygenase
- ICAM
- intercellular adhesion molecule
- PMN
- polymorphonuclear leucocyte
- VCAM
- vascular cell adhesion molecule
- TNF
- tumour necrosis factor