Article Text

Abstract
BACKGROUND Inflammatory bowel disease (IBD) is characterised by infiltration of inflamed mucosal regions with CD4+ T lymphocytes and other mononuclear cells. Interleukin (IL)-16 exerts a strong chemoattractant activity on CD4+ cells. Moreover, IL-16 activates expression and production of proinflammatory cytokines such as IL-1β, IL-6, IL-15, and tumour necrosis factor α (TNF-α) in human monocytes.
AIM To examine if IL-16 expression is increased in IBD patients compared with healthy controls.
METHODS Twenty one patients with IBD (10 with ulcerative colitis (UC), 11 with Crohn's disease (CD)), seven disease specificity controls (DSC), and seven healthy controls were studied. Biopsies were taken during colonoscopies and IL-16 mRNA as well as protein expression were investigated by reverse transcriptase-polymerase chain reaction, ELISA, western blot, and immunohistochemistry.
RESULTS IL-16 mRNA and protein expression in the colonic mucosa of IBD patients were increased twofold compared with healthy controls, DSC, or IBD patients under steroid treatment. Most of the detected IL-16 protein was in its bioactive 17 kDa form and was predominantly expressed in eosinophils. Increased IL-16 expression in UC patients appeared to be mainly restricted to the inflamed regions of the colonic mucosa. Levels of caspase 3, which processes the 68 kDa IL-16 precursor molecule into the biological active 17 kDa form, were not increased.
CONCLUSIONS Our results provide evidence that IL-16 expression is significantly increased in the inflamed colonic mucosa of IBD patients but not in control individuals, DSC, or patients under steroid treatment. Therefore, upregulation of IL-16 expression seems to be specific for chronic intestinal inflammation and could lead to increased secretion of other proinflammatory cytokines in IBD.
- interleukin-16
- T lymphocytes
- eosinophils
- Crohn's disease
- ulcerative colitis
- inflammatory bowel disease
Abbreviations used in this paper
- CD
- Crohn's disease
- UC
- ulcerative colitis
- IBD
- inflammatory bowel disease
- RT-PCR
- reverse transcriptase-polymerase chain reaction
- TNF-α
- tumour necrosis factor α
- IFN-γ
- interferon γ
- DSC
- disease specificity controls
- BSA
- bovine serum albumin
- PBS
- phosphate buffered saline
- anti-EOP
- anti-eosinophilic peroxidase
Statistics from Altmetric.com
Interleukin (IL)-16, formerly known as lymphocyte chemoattractant factor, was first described in 1982.1 ,2It is secreted by CD8+ peripheral blood lymphocytes,3-5 eosinophils,6 mast cells,7 and non-immune epithelial cells8after stimulation with mitogens, antigens, or vasoactive amines—for example, histamine.9 Although it has been shown that IL-16 requires expression of CD4 on target cells to induce a migratory signal,10 a recent study demonstrated that functional activity of IL-16 can be independent of CD4.11 This indicates the existence of another receptor for IL-16 apart from CD4. In addition to its properties as a chemoattractant for CD4 expressing cells, it stimulates production of proinflammatory cytokines such as IL-6, tumour necrosis factor α (TNF-α), IL-1β, and IL-15 by monocytes,12 upregulation of IL-2 receptor alpha (IL-2Rα) and IL-2Rβ on T cells,13 and initiates the stress pathway by activation of the stress activated protein kinase and p38 mitogen activated protein kinase.14
Nevertheless, the biological role of IL-16 in vivo remains uncertain. One hypothesis is that it may contribute to antigen independent non-clonal recruitment and priming of CD4+ cells in inflammatory processes. In asthma, large numbers of activated CD4+ cells can be found in the airway mucosa.15 IL-16 immunoreactivity as well as IL-16 mRNA expression are elevated in bronchial biopsies obtained from asthmatic subjects compared with normal non-asthmatic controls. A high correlation between the amount of detectable IL-16 protein and mRNA and number of infiltrating CD4+ lymphocytes was demonstrated. These studies suggest that in asthma, the airway epithelium is induced to produce IL-16. Because of its ability to induce lymphocyte migration and to activate T cells for proliferation, IL-16 can be classified as a proinflammatory cytokine.
Crohn's disease (CD) and ulcerative colitis (UC) are characterised by an increase in proinflammatory cytokine and chemokine expression (for example, IL-1β, IL-12, TNF-α, interferon (IFN)-γ, IL-6, IL-8, RANTES, MCP-1) as well as infiltration by CD4+ cells into the inflamed regions of the intestinal mucosa.16-19 In this study we investigated if IL-16 protein and mRNA expression were elevated in inflammatory bowel diseases (IBD). We demonstrated that in colonic biopsies obtained from inflammatory lesions of patients with active CD and UC, IL-16 mRNA expression was increased compared with normal controls, as demonstrated by reverse transcriptase-polymerase chain reaction (RT-PCR) assays. A similar increase in IL-16 protein was detected in comparison with normal controls, disease specificity controls (DSC), or IBD patients treated with corticosteroids. Recently it was described that IL-16 is produced as a 68 kDa precursor protein which is cleaved by caspase 3 into the biologically active 17 kDa form.20 Our study demonstrates that the predominant form of IL-16 in the inflamed colonic mucosa is the 17 kDa molecule. Moreover, increased IL-16 expression (precursor and active form) in UC patients is mainly restricted to inflamed mucosal regions of the colon. In parallel, we show that caspase 3 mainly exists in its inactive 32 kDa form and is not over expressed in IBD patients compared with controls. This indicates that caspase 3 does not appear to be responsible for higher IL-16 levels in IBD. Our results provide evidence that IL-16 is over expressed in the inflamed colonic mucosa of IBD patients and may therefore contribute to the inflammatory processes in IBD by recruiting CD4+ cells to inflamed colonic areas and by enhancing expression of other proinflammatory cytokines such as TNF-α, IL-1β, and IL-6.
Material and methods
PATIENTS
A total of 21 patients with IBD (10 patients with UC and 11 patients with CD), seven disease specificity controls (DSC; patients with diverticulitis, salmonellosis, or infectious enterocolitis), and seven controls participated in this study (table 1). All IBD patients (including steroid treated) were endoscopically active with a Crohn's disease activity index21 ranging from 154 to 402 (median 204) or a colitis activity index22 ranging from 5 to 12 (median 8.9), respectively. Six of 10 UC patients and 8/11 CD patients received treatment with corticosteroids (prednisone or budesonide). Eight of 10 UC patients and 10/11 CD patients were also treated with oral salicylates (mesalazine, salasulphapyridine). In addition, one CD patient received 5-ASA and another CD patient was treated with IFN-β. None of the patients was treated with immunosuppressives (for example, azathioprine). Diagnosis had been established by clinical, endoscopic, histological, and/or radiological criteria. All IBD patients underwent sigmoidoscopy or colonoscopy for routine clinical evaluation. Controls included samples from seven patients with irritable bowel syndrome.
Clinical data for patients with Crohn's disease (CD) and ulcerative colitis (UC)
REAGENTS
Mouse antihuman CD4 monoclonal antibody, mouse anti-IL-16 monoclonal capture antibody, and biotinylated rabbit anti-IL-16 polyclonal detection antibody were purchased from Pharmingen (Hamburg, Germany). Cy3 conjugated goat antimouse antibody and biotin-SP conjugated ChromePure Rabbit IgG were from Jackson Immuno Research Laboratories Inc. (West Grove, Pennsylvania, USA). Rabbit anticaspase 3 polyclonal antibody was from Upstate Biotechnology (Lake Placid, USA). Murine anti-eosinophil peroxidase monoclonal antibody was purchased from Biotrend (Cologne, Germany).
RNA ISOLATION AND RT-PCR
Total RNA from biopsies was obtained using a RNeasy kit from Qiagen (Hilden, Germany) according to the manufacturer's manual. Briefly, one frozen biopsy was homogenised under liquid nitrogen, resuspended in 600 μl of lysis buffer RLT, passed five times through a 21 gauche needle, and centrifuged for three minutes at 20 000g; 600 μl of 70% ethanol were added and the sample applied in 600 μl portions onto an RNeasy spin column. The column was centrifuged for 15 seconds at 8000g and the flow through discarded. The column was washed with 700 μl of wash buffer RW1 and centrifuged as described previously; 500 μl of wash buffer RPE were added and the column again centrifuged. After addition of another 500 μl of RPE, the column was centrifuged for two minutes at 20 000g. RNA was eluted by addition of 50 μl of bidistilled sterile water (supplemented with 0.1% DEPC) and a final centrifugation at 8000 g. Total RNA amounts were spectrometrically determined and samples were stored at −80°C until use. The quality of isolated RNA was analysed on agarose gels under standard conditions.
For RT-PCR, 1 μg of total RNA was reverse transcribed to cDNA using the Advantage RT-for-PCR kit from Clontech (Palo Alto, California, USA), according to the manufacturer's protocol. For amplification of IL-16 cDNA, 5 μl of a 1:5-fold diluted RT mix were added to 1× PCR buffer (Perkin Elmer, Foster City, California, USA), 0.2 μM of each primer, 0.5 μM dNTP mix, 1 U Taq polymerase (Perkin Elmer), and 1.5 mM MgCl2 in a total volume of 50 μl. The reaction was immediately started by a denaturation step for five minutes at 94°C, followed by 32 cycles of 20 seconds at 94°C, 30 seconds at 67°C, and 90 seconds at 72°C, and a final extension step for seven minutes at 72°C. The desired length of the PCR product which was finally analysed on a 1% agarose gel was 748 bp. As a control, β-actin cDNA was amplified in parallel experiments. The cycling programme was five minutes at 94°C, 33 cycles of 30 seconds at 94°C, 20 seconds at 60°C, and 90 seconds at 72°C, followed by a final extension step for seven minutes at 72°C. The purity of all RNA samples and specificity of the reactions was checked by performing PCR experiments as described without prior reverse transcription. All primers were purchased from Roth (Karlsruhe, Germany). IL-16 (sense): 5′-GAT ACC ACA GCC GAA GAC CCT TGG-3′; IL-16 (antisense): 5′-GTG CTC GCT GGC TAG GCA TCT TG-3′; β-actin (sense): 5′-TGT GAT GGT GGG AAT GGG TCA-3′; β-actin (antisense): 5′-TTT GAT GTC ACG CAC GAT TTC C-3′.
IL-16 ELISA
One biopsy from each patient was homogenised under liquid nitrogen and resuspended in 150 μl of RPMI medium without serum. The solution was briefly sonicated, and the protein concentration determined and adjusted to 0.5 mg/ml. This solution (50 μl) was used in an ELISA as follows: a microtitre plate (Nunc, Maxisorb) was coated overnight at 4°C with 50 μl of primary mouse antihuman IL-16 monoclonal capture antibody diluted 1:250 in carbonate buffer (200 mM sodium bicarbonate, 80 mM sodium carbonate, pH 9.5). The plate was washed three times with phosphate buffered saline (PBS) with 0.1% Tween 20 (PBST) and blocked for one hour with 100 μl of PBST supplemented with 0.5% bovine serum albumin at room temperature. After washing for three times with PBST, standards ranging from 10 to 0.08 ng/ml or 50 μl of each sample (corresponding to 25 μg of total protein) were added to the wells and incubated for two hours at room temperature. The plate was washed six times with PBST, and 50 μl of biotinylated rabbit antihuman/mouse IL-16 polyclonal detection antibody diluted 1:250 in PBST were added. After incubation for one hour at room temperature and washing six times with PBST, 50 μl of ExtrAvidin-peroxidase (Sigma) diluted 1:500 were added and incubated for 45 minutes at room temperature. The plate was washed six times with PBST and 50 μl of OPD solution (4 mgo-phenylenediamine-dihydrochloride (Sigma) dissolved in 1 ml of 20 mM citric acid, 50 mM monobasic sodium phosphate, pH 5.0; 5 ml of this solution were freshly supplemented with 3 μl of 30% hydrogen peroxide). The reaction was stopped by addition of 100 μl of 1 M HCl and plates were read at 490 nm.
TISSUE PROCESSING AND IMMUNOFLUORESCENCE
Biopsies were embedded in cryomatrix and snap frozen in liquid nitrogen. Thick cryostat sections (7 μm) were thaw mounted on Superfrost slides (Scientific Supply Source, Aurora, USA) postfixed for five minutes in acetone, air dried, and stored at −20°C before staining. The tissues in the slides were permeabilised with 0.1% Triton X-100 in 0.1 M PBS, washed three times in PBS, and blocked in 0.75% bovine serum albumin (BSA) in PBS. For double staining with anti-IL-16 and anti-CD4 or anti-eosinophilic peroxidase (anti-EOP) antibody, respectively, sections were first incubated for one hour with monoclonal mouse antihuman CD4 antibody or anti-EOP (diluted 1:200 in 0.75% BSA in PBS), washed three times with PBS, blocked for 20 minutes in 0.75% BSA in PBS, and then incubated with biotinylated rabbit antihuman/mouse IL-16 (diluted 1:200 in 0.75% BSA in PBS) for one hour at room temperature. After washing in PBS, tissue bound antibodies were detected as follows: CD4 was detected by incubation with Cy3 conjugated goat antimouse antibody (Jackson Immuno Research Laboratories, West Grove, Pennsylvania, USA) diluted 1:150 in 0.75% BSA/PBS. The IL-16 antibody was detected by avidin-FITC (Vector) diluted 1:100 in 5% human serum. Fluorescence was visualised using an Axiophot microscope (Zeiss, Germany) with the appropriate filter systems and photos were taken on Provia 1600 colour films (Fuji).
WESTERN BLOT
IL-16 as well as caspase 3 were detected in colonic biopsies by western blot analysis as follows. One biopsy was homogenised under liquid nitrogen, resuspended in 300 μl of lysis buffer (1% SDS, 10 mM Tris (pH 7.4), 1% phosphatase inhibitor cocktail (Sigma)), agitated for one hour at 4°C, and sonicated for five seconds. The lysate was centrifuged at 16 000 g and 4°C for 15 minutes and the supernatant was transferred to a fresh tube. Total protein (20 μg) was separated on a 12% polyacrylamide gel under standard conditions. Proteins were transferred to PVDF membrane (Amersham Pharmacia, Uppsala, Sweden) by semi dry blotting (Bio Rad, Munich, Germany) and the membrane blocked for one hour at room temperature with 3% dry milk in TTBS (20 mM Tris (pH 7.6), 0.137 M NaCl, 0.1% Tween-20). Anti-IL-16 antibody (Pharmingen) diluted 1:200 or 2 μg/ml of anti-caspase 3 antibody (Upstate Biotechnology), respectively, were added and the membrane incubated overnight at 4°C under agitation. As secondary antibody, horseradish peroxidase conjugated antimouse or antirabbit antibodies, respectively, were used at a dilution of 1:2000 and detected with an ECL Plus kit (Amersham Pharmacia).
STATISTICAL ANALYSIS
Results are expressed as mean (SD) and p<0.05 was accepted as statistically significant.
Results
IL-16 mRNA EXPRESSION IS ELEVATED IN COLONIC BIOPSIES FROM PATIENTS WITH IBD
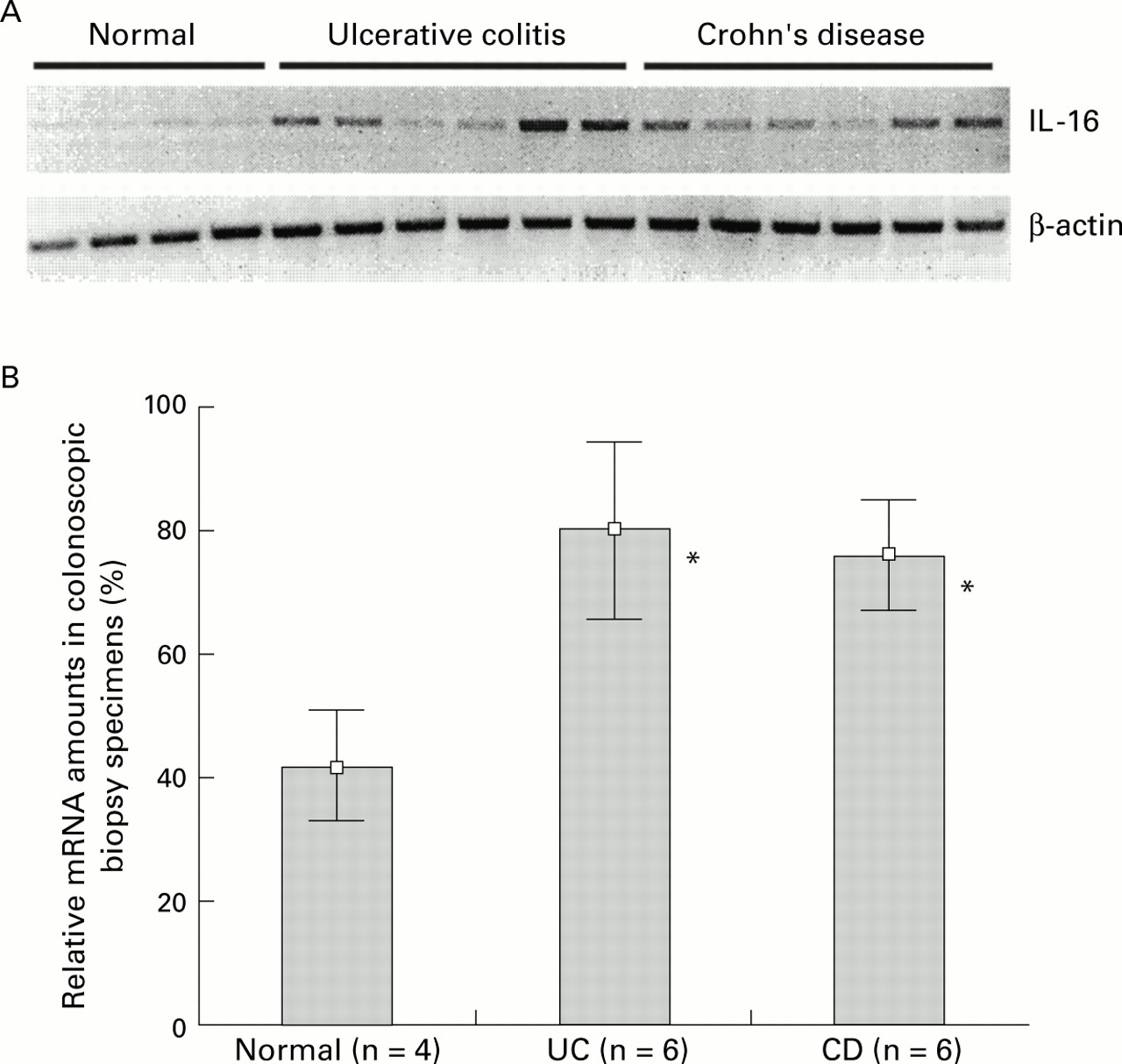
Total RNA was extracted from biopsies from four control individuals as well as from 12 patients with IBD (six UC and six CD patients). IL-16 and β-actin specific cDNA amplifications were performed as described. All PCR products were analysed on a 1% agarose gel (fig 1A, top). β-actin cDNA was amplified in a parallel experiment to document the use of identical RNA amounts in all samples (fig 1A, bottom). The number of cycling reactions necessary to get into the linear amplification range was determined in separate experiments (data not shown). In contrast with the control samples (lanes 1–4), the amount of IL-16 specific PCR products was markedly elevated in biopsies from UC (lanes 5–10) and CD (lanes 11–16) patients. The quantity of the products was determined densitometrically using SigmaGel (version 1.0, Jandel Scientific, Chicago, USA). The density of the band with the highest strength (lane 9) was set to 100% during densitometric evaluation and remaining lanes were normalised to this value. Results are shown in fig 1B as mean (SD). IL-16 mRNA expression was significantly (p<0.05) increased about twofold in UC (mean 79.59; range 63.75–100) and CD (mean 75.1; range 61.21–86.23) colonoscopic biopsy specimens in comparison with controls (mean 41.9; range 31.41–51.01).

(A) Total RNA was extracted from biopsies from four normal control individuals and 12 patients with IBD (six Crohn's disease and six ulcerative colitis patients). Interleukin 16 (IL-16) mRNA was amplified by reverse transcriptase-polymerase chain reaction (RT-PCR) and analysed on an agarose gel (top panel). The use of equal amounts of total RNA was confirmed in a parallel experiment by amplification of β-actin mRNA (bottom panel). PCR products were analysed on a 1% agarose gel. (B) Densitometric quantification of IL-16 specific PCR products. Values are given as mean (SD) percentage of intensity of the highest detected densitometric strength. *p<0.05 v controls.
LEVELS OF IL-16 PROTEIN ARE INCREASED IN COLONOSCOPIC BIOPSY SPECIMENS FROM IBD PATIENTS
To determine if elevated expression of IL-16 mRNA in IBD was related to an increase in IL-16 protein production, biopsies from seven controls, seven DSC, and 21 patients with IBD were homogenised under liquid nitrogen and proteins were extracted as described in the methods section. The amount of total protein (ranging from 120 to 160 μg/mg tissue) was not increased in IBD biopsies compared with DSC or normal controls. IL-16 was detected by ELISA. Results are shown in fig 2. In both active UC (mean 14.76; range 8.46–20.51) and CD (mean 13.98; range 8.51–21.39), IL-16 protein (ng/ml of biopsy lysate) was significantly (p<0.05) increased about twofold in comparison with controls (mean 6.31; range 5.45–9.72), DSC (mean 5.63; range 2.14-8.69), steroid treated patients with UC (mean 6.54; range 3.39–11.84), and CD (mean 9.94; range 3.72–15.14). Our data indicate that elevated IL-16 mRNA expression in IBD shown in fig 1 relates to an increase in IL-16 protein production in IBD.

Quantification of interleukin 16 (IL-16) protein in colonic mucosal biopsy specimens from 21 patients with inflammatory bowel disease (10 ulcerative colitis (UC) patients (6/10 under corticosteroid (cs) therapy), 12 Crohn's disease (CD) patients (8/12 under cs treatment)), seven disease specificity controls (DSC), and seven normal individuals. Tissues were lysed and adjusted to a protein concentration of 0.5 mg/ml: 50 μl of each sample were used to detect IL-16 content by IL-16 specific ELISA. Values are mean (SD).
DETECTION OF IL-16 IN BIOPSIES FROM IBD PATIENTS BY IMMUNOFLUORESCENCE STAINING
To determine if infiltration of inflamed mucosal tissue by CD4+ lymphocytes correlated with an increase in IL-16 protein, biopsies from control individuals and patients with IBD were double stained with specific fluorescence labelled anti-CD4 and anti-IL-16 antibodies. The specificity of the IL-16 antibody was controlled in parallel experiments under the same conditions with an irrelevant isotype antibody (fig 3, right). Figure 3 shows representative results obtained from one of 3–4 identical experiments. CD4 and IL-16 positive cells were detected in the control tissue only at low frequencies (fig 3, top panel). In contrast, a marked increase in CD4+ cells as well as IL-16 expressing cells was seen in samples from CD (fig 3, middle panel) and UC (fig 3, bottom panel) patients. In further experiments, stainings with a monocyte/macrophage specific anti-CD68 antibody (Ki-M6) and an anti-CD8 antibody (Dianova, Germany) were performed, but no significantly increased amounts of the tested cells in IBD specimens were detected compared with controls. These results confirm our previous data, and taken together indicate that IL-16 expression is significantly increased in inflamed mucosal regions of IBD patients.

Detection of CD4 and interleukin 16 (IL-16) by immunofluorescence staining in biopsies from control individuals and patients with inflammatory bowel disease (ulcerative colitis and Crohn's disease). The specificity of the IL-16 antibody was confirmed by an irrelevant isotype antibody (biotin conjugated rabbit IgG). CD4 infiltration was visualised with a Cy3 conjugated antibody (red) while IL-16 protein was detected by an FITC labelled antibody (green).
Only some of the cells which were stained with the anti-CD4 antibody were also detected by the anti-IL-16 antibody, indicating that colonic mucosal IL-16 is predominantly produced by cells different from CD4+ cells. Therefore, another set of double stainings with anti-IL-16 and eosinophil specific anti-EOP was performed. Figure 4demonstrates that most of the cells which were IL-16 positive also showed staining with anti-EOP. This double positivity suggests that eosinophils strongly contribute to expression and secretion of IL-16 in IBD.
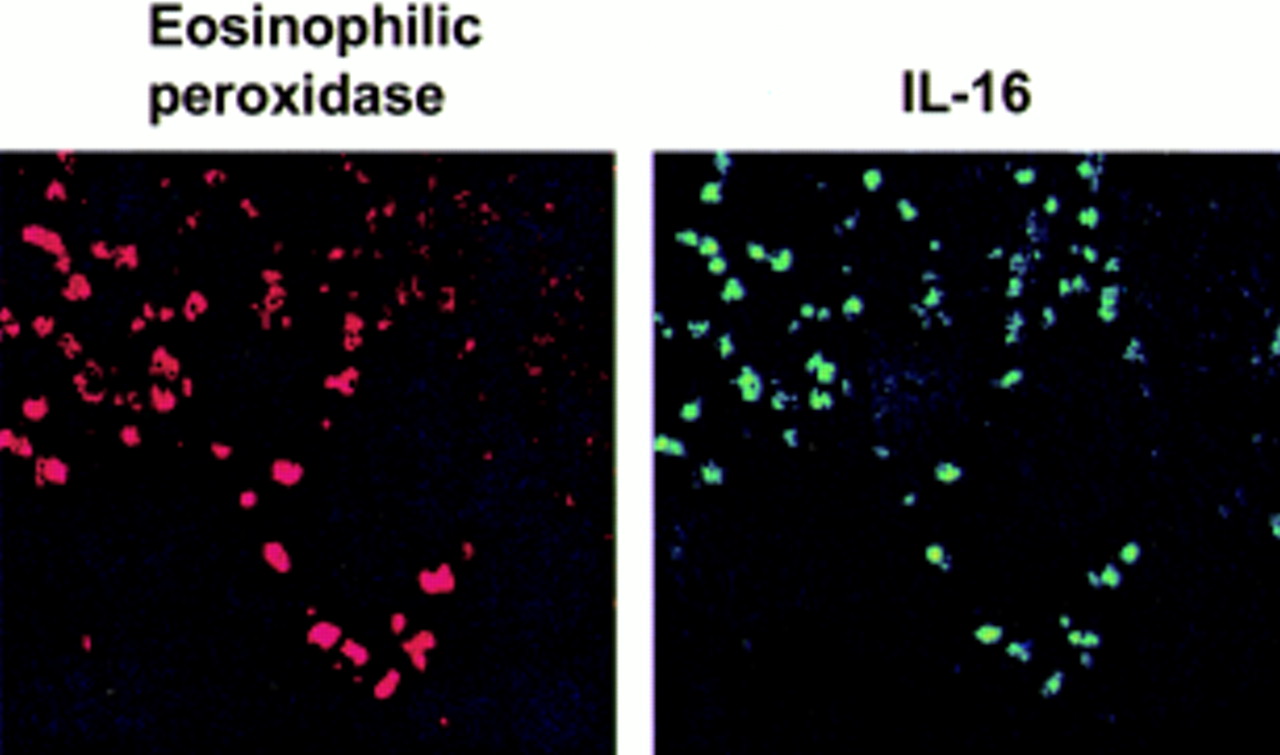
Staining of biopsies from Crohn's disease patients with anti-eosinophilic peroxidase antibody (left) and anti-interleukin 16 (IL-16) antibody (panel).
IL-16 IN IBD BIOPSIES PREDOMINANTLY EXISTS IN ITS BIOACTIVE 17 kDa FORM
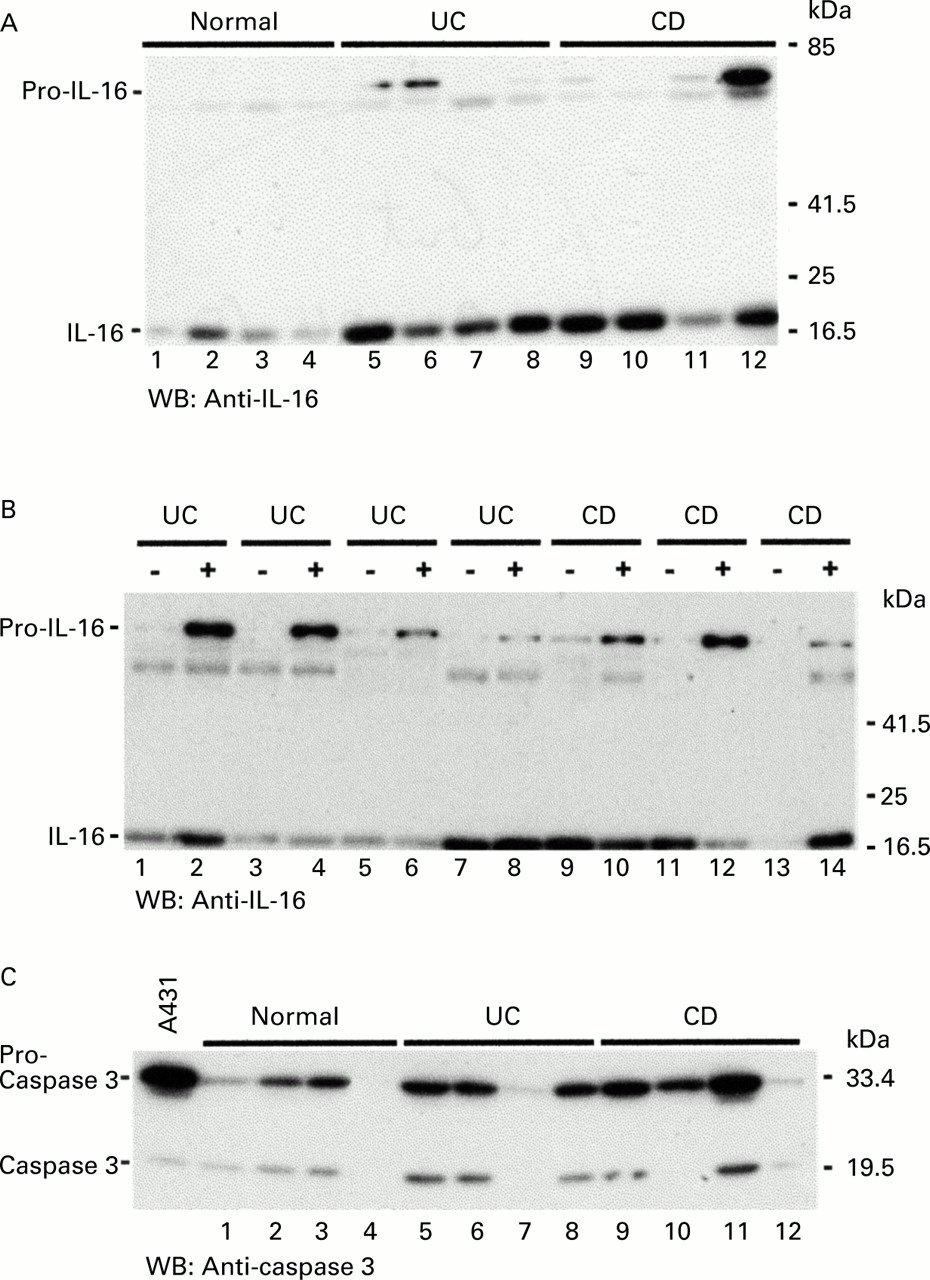
To investigate if IL-16 detected in biopsies from patients with IBD was bioactive, specimens from four normal controls, four UC patients, and four CD patients were extracted to assess if IL-16 was present in an inactive 68 kDa precursor form (pro-IL-16) or the processed biologically active 17 kDa form. Lysates were analysed on polyacrylamide gels and IL-16 detected in western blot assays. The results are shown in fig 5A. As already demonstrated by RT-PCR and ELISA, IL-16 expression was significantly upregulated in colonic samples from CD and UC patients (lanes 5–12) compared with normal controls (lanes 1–4). Moreover, pro-IL-16 was detected in two UC patients (lanes 5, 6) and one CD patient (lane 12) in large amounts. Next we investigated if increased IL-16 amounts could also be detected in non-inflamed regions of the colon of IBD patients (fig 5B). Biopsies from either non-inflamed (−) or inflamed (+) colonic mucosa were taken from four UC and three CD patients and tested in western blot experiments. Although one UC patient (lanes 7, 8) showed similar IL-16 amounts in both inflamed and non-inflamed tissue samples, it appeared that in three of four UC patients IL-16 production was increased in the inflamed colonic mucosa. The outcome for CD patients was more inhomogeneous. In one individual IL-16 expression was increased in the inflamed tissue (lanes 13, 14) and two others showed similar amounts of IL-16 in uninflamed and inflamed specimens (lanes 9–13). This experiment indicates that IL-16 is found mainly in specimens from inflamed colonic regions in UC patients, whereas IL-16 production in CD patients does not appear to be restricted to inflamed mucosal regions.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Interleukin 16 (IL-16) protein expression was analysed by western blotting in biopsies taken from four control individuals (lanes 1–4), four ulcerative colitis (UC) patients (lanes 5–8), and four Crohn's disease (CD) patients (lanes 9–12). The experiment revealed the predominant existence of the bioactive 17 kDa form of IL-16 in IBD patients compared with controls. (B) Biopsies from four UC and three CD patients were taken from non-inflamed (—) or inflamed (+) regions of the colon and tested for IL-16 protein expression in a western blot assay. (C) Extracts of the same biopsies as in (A) were used to detect caspase 3 protein expression. A lysate from unstimulated human A431 cells supplied with the anti-caspase 3 antibody served as a control (WB, western blot).
Because it has recently been described that pro-IL-16 is processed into the 17 kDa molecule by caspase 3,20 we investigated if there were differences in caspase 3 levels between normal control biopsies and samples from IBD patients. The same samples as those in fig 5A were used for caspase 3 specific western blots. Figure 5C demonstrates that similar amounts of caspase 3 were expressed in controls as in IBD specimens. Interestingly, the main form of caspase 3 was its inactive 32 kDa form (pro-caspase 3). Three samples (lanes 4, 7, 12) appeared not to contain caspase 3 protein within the detection limit of our assay. Samples with higher amounts of active caspase 3 (fig 5C, lanes 6 and 11) did not necessarily contain high levels of active IL-16 (fig 5A, lanes 6 and 11). This suggests that increased caspase 3 protein expression does not seem to be responsible for increased amounts of IL-16 protein in inflamed colonic mucosal tissue.
Discussion
There is strong evidence that inflammatory immunoregulation in the intestinal mucosa is characterised by increased concentrations of proinflammatory cytokines and chemokines, including IL-1β, TNF-α, IFN-γ, IL-6, IL-8, IL-12, RANTES, or MCP-1.18 ,19 ,23-28 These molecules play a major role in intestinal inflammation by recruiting and activating inflammatory cells. IL-16 is a cytokine that is known to be a chemoattractant for CD4+ T lymphocytes.1 ,2 Moreover, IL-16 stimulates expression and production of proinflammatory cytokines such as TNF-α, IL-1β, IL-6, and IL-15 in human monocytes.12It has recently been described that large numbers of CD4+and CD8+ cells infiltrate the inflamed intestinal mucosa in patients with active IBD.17 Therefore, the aim of this study was to investigate if IL-16 activity was elevated in patients with CD or UC. We demonstrated that not only IL-16 mRNA (fig 1) but also IL-16 protein (figs 2, 3, 5A, 5B) were significantly elevated in the inflamed colonic mucosa of patients with UC and CD. According to its biological properties we hypothesise that IL-16 could be involved in the recruitment of CD4+ lymphocytes into inflamed mucosal tissues and in addition could contribute to increased expression of other proinflammatory cytokines in IBD.
IL-16 exists as a 68 kDa precursor molecule (pro-IL-16) specifically processed by caspase 3 into a 17 kDa molecule, which constitutes the bioactive secreted form of IL-16.20 ,29 CD8+and CD4+ T lymphocytes constitutively express IL-16 mRNA and produce pro-IL-16 protein.30 ,31 But although bioactive IL-16 is detectable in unstimulated CD8+ T cells, it is not present in resting CD4+ T cells. In contrast, activated CD4+ T cells secrete bioactive IL-16.32 We used immunohistology to determine the source of IL-16 in the inflamed colonic mucosa. We found that only a small part of the cells that were expressing the surface marker CD4 were also stained by the anti-IL-16 antibody (fig 3). In contrast, an antibody specific for eosinophilic peroxidase detected most of the cells which also expressed IL-16 (fig 4). Our results suggest that a small percentage of IL-16 protein in the inflamed colonic mucosa could be produced by CD4+ cells but that the majority of IL-16 is secreted by eosinophils.
Most of the IL-16 protein detected in the ELISA experiments (fig 2) consisted of the 17 kDa form, as determined by parallel western blots (fig 5A). Furthermore, increased IL-16 expression was mainly observed in biopsies from inflamed mucosal regions from UC patients (fig 5B). In contrast, two of three CD patients contained similar amounts of IL-16 in inflamed and non-inflamed colonic tissues. One might hypothesise that increased amounts of bioactive IL-16 may be caused by elevated expression or activity of caspase 3. But as we have shown here, there were no significant differences in caspase 3 expression between controls and IBD specimens. Moreover, the large amounts of caspase 3 detected in colonic mucosal biopsies was in its inactive 32 kDa form (fig 5C). Therefore, it is likely that other molecular mechanisms or molecules are responsible for this phenomenon, such as changes in the regulatory mechanisms of the IL-16 gene or variations in IL-16 transcription and/or translation rates. Experiments to address the transcriptional regulation of the IL-16 gene are currently in progress.
Taken together our results provide evidence for increased expression of IL-16 in IBD that appears to be restricted to inflamed colonic regions in UC patients. Although our experiments revealed “only” a twofold increase in IL-16 expression in IBD patients, this could be enough to cause a marked inflammatory response because IL-16 is described as a potent chemoattractant. Therefore, IL-16 could contribute to the inflammatory processes in IBD by promoting recruitment and activation of inflammatory CD4+ cells1 ,2 and by inducing expression of other important proinflammatory cytokines. Previous studies showed that anti-CD4 antibody treatment of CD patients or mice with TNBS colitis resulted in beneficial effects for both diseases.33 ,34 Because IL-16 also uses CD4 as a receptor, IL-16 specific inhibitors could have therapeutic implications in suppressing CD4+ T cell mediated inflammation of the intestine.
Acknowledgments
This work was supported by research fellowship from Boehringer Ingelheim (DS), the Deutsche Forschungsgemeinschaft (Sch 512/1–3), SFB415, and MFG. Parts of the work have been presented to the annual meeting of the American Gastroenterological Association (AGA), May 1999, Orlando, Florida, and were published in abstract form. We would like to thank Kirstin Schirrmacher and Brigitte Mauracher for exellent technical assistance.
Abbreviations used in this paper
- CD
- Crohn's disease
- UC
- ulcerative colitis
- IBD
- inflammatory bowel disease
- RT-PCR
- reverse transcriptase-polymerase chain reaction
- TNF-α
- tumour necrosis factor α
- IFN-γ
- interferon γ
- DSC
- disease specificity controls
- BSA
- bovine serum albumin
- PBS
- phosphate buffered saline
- anti-EOP
- anti-eosinophilic peroxidase