Article Text

Abstract
Coeliac disease (CD) is caused by a CD4 T helper cell type 1 (Th1) response in the small intestinal mucosa to dietary gluten. As the major Th1 inducing cytokine, interleukin 12, is undetectable in CD gut mucosa, the mechanism by which Th1 effector cells are generated remains unknown. Interferon (IFN) α, a cytokine capable of promoting IFN-γ synthesis, has been implicated in the development of Th1 mediated immune diseases. Here we report a case of CD-like enteropathy in a patient receiving IFN-α for chronic myeloid leukaemia. Morphological assessment of duodenal biopsies taken from the patient showed total villous atrophy, crypt cell hyperplasia, and a high number of CD3+ intraepithelial lymphocytes. Both antigliadin antibodies and antiendomysial antibodies were positive. RNA analysis revealed pronounced expression of IFN-γ. Withdrawal of gluten from the diet resulted in a patchy improvement in intestinal morphology, normalisation of laboratory parameters, and resolution of clinical symptoms. By western blot analysis, IFN-α protein was seen in the duodenal mucosa from untreated CD patients but not in controls. This was associated with marked expression of IFN-γ protein in CD mucosa. Collectively, these results suggest a role for IFN-α in promoting Th1 responses to gluten.
- coeliac disease
- interferon
- small intestine
- T helper cell response
Abbreviations used in this paper
- CD
- coeliac disease
- EMA
- antiendomysial antibodies
- AGA
- antigliadin antibodies
- Th1,T helper cell type 1
- IFN, interferon
- IL
- interleukin
- IEL
- intraepithelial lymphocyte
- APC
- antigen presenting cell
Statistics from Altmetric.com
Coeliac disease (CD) affects the proximal small intestine and is caused by a local immune response to dietary gluten in genetically susceptible individuals. The characteristic features of CD inflammation are villus atrophy, crypt cell hyperplasia, and increased number of intraepithelial lymphocytes (IEL). Mucosal surface area is lost as the villi become shorter, causing malabsorption and diarrhoea.1
A growing body of evidence indicates that CD4+ T cell mediated hypersensitivity plays a major role in tissue injury in CD. Lamina propria CD4+ T cells are phenotypically activated and produce large amounts of proinflammatory cytokines when exposed to gluten.2-4 Previous experimental studies have also shown that direct activation of lamina propria T cells in explant cultures of human fetal gut produces villous atrophy and crypt cell hyperplasia.2 ,5 ,6 These pathological changes are associated with increased expression of T helper cell type 1 (Th1) cytokines, such as interferon (IFN) γ and tumour necrosis factor α.2 ,5 ,6 However, there is also increasing evidence that mucosal remodelling, which is seen in CD, is due to cytokine induced perturbations in the production of matrix degrading enzymes and epithelial growth factors by lamina propria fibroblasts.2 ,5-7
Differentiation of T cells towards the Th1 or Th2 subtype is influenced by many factors, including the nature and concentration of the antigen, type of antigen presenting cell (APC), and polarising signals present in the microenvironment.8 Paradoxically, the major Th1 inducing factor, interleukin (IL) 12, is virtually undetectable in the gut mucosa of patients with CD, despite the clear polarisation of T cell responses to gluten along the Th1 pathway.3 The mechanism by which Th1 effector cells are generated in CD mucosa therefore remains unknown.
IFN-α is a cytokine produced by virally infected cells. One of its main immunomodulatory activities is its ability to induce and maintain Th1 cells.9 There is a strong correlation between IFN-α expression and development of autoimmune diseases in humans and experimental animals.9-11
In this study we first describe a case of CD, responsive to gluten elimination, in a patient receiving IFN-α treatment for chronic myeloid leukaemia. We also show elevated IFN-α in mucosal biopsies of patients with untreated CD, suggesting a role for IFN-α in promoting Th1 responses to gluten.
Methods
PATIENTS AND CONTROLS
Three biopsy specimens from the distal duodenum of eight patients with untreated CD (aged 5–13 years) were obtained during upper gastrointestinal endoscopy. One specimen was used for routine histological examination whereas the remaining two were immediately frozen in liquid nitrogen and stored until tested. The delay between the beginning of abdominal symptoms and collection of biopsies was 3–12 months. No patient had a recent history of clinically evident intestinal infection. The histopathological diagnosis was based on typical mucosal lesions with crypt cell hyperplasia, villous atrophy, and increased number of IELs. All CD patients were positive for antiendomysial (EMA) and antigliadin (AGA) antibodies. In no patient was gluten challenge performed to confirm the CD diagnosis. A gluten free diet resulted in resolution of symptoms and disappearance of both EMA and AGA after 4–12 months. Control patients (n=5; aged 4–16) were under investigation for gastrointestinal symptoms but had normal histology and were EMA and AGA negative. Biopsy specimens were snap frozen in liquid nitrogen and stored at −70°C until used.
IMMUNOHISTOCHEMISTRY
Frozen sections (6 μm) were stained with anti-CD3 antibody using the indirect peroxidase method.5 ,6
RNA EXTRACTION, CDNA PREPARATION, AND CYTOKINE RNA DETERMINATION
RNA was extracted from biopsy specimens using 1 ml of a monophasic solution of phenol and guanidine isothiocyanate (TRIZOL, Life Technologies, Paisley, UK) and chloroform, followed by isopropanol (Sigma Chemical, Co, St Louis, Missouri, USA) precipitation. Complementary DNA (cDNA) was synthesised from 1 μg of total RNA, as previously described.12 Transcripts for IFN-γ were determined by Southern blotting.12
WESTERN BLOT ANALYSIS
Total protein was extracted from mucosal samples of CD patients and controls as previously reported.12 For detection of IFN-α, 300 μg of total protein was separated using 15% sodium dodecyl sulphate-polyacrylamide gel electrophoresis. IFN-α was detected after incubation with a rabbit antihuman IFN-α polyclonal antibody (1:100 final dilution; Immunokontact, Frankfurt, Germany) and subsequent incubation with a goat antirabbit IgG monoclonal antibody conjugated with peroxidase. After detection of IFN-α by enhanced chemiluminescence, blots were stripped by incubation for 30 minutes at 50°C in stripping medium (2% sodium dodecyl sulphate, 0.05 M Tris (pH 6.8), 0.1 mM β-mercaptoethanol), and subsequently incubated with a goat antihuman IFN-γ polyclonal antibody (Santa Cruz Biotechnology Inc., Santa Cruz, California, USA). IFN-γ protein was identified as above.
Case report
A 61 year old woman presented in May 1999. She had a history of chronic myeloid leukaemia (Philadelphia chromosome positive) diagnosed in June 1998. At the time of diagnosis she was found to have leucocytosis, anaemia, and thrombocytosis. Following initial treatment with hydroxyurea, she was entered into a controlled trial and randomised to treatment with high dose IFN-α 10 MU/day (Wellferon) subcutaneously, in September 1998. Before the start of treatment, she was clinically well with no abdominal or bowel symptoms. She weighed 91 kg (body mass index 33.8 kg/m2) and had been overweight all of her adult life. She had no past history of note and there was no family history of autoimmune disease. Urea and electrolytes, liver biochemistry, B12 and red cell folate, and C reactive protein were within the normal range. The patient was reviewed regularly in the haematology clinic but felt unwell from the onset of IFN-α therapy, complaining of fatigue, anorexia, and loose bowel movements. During that time no other drug was administered. In December 1998, three months after starting treatment, a 7 kg weight loss was documented and symptoms persisted. In January 1999, the dose of IFN-α was reduced to 7 MU daily. During early 1999 she continued to experience worsening diarrhoea which did not respond to antidiarrhoeal therapy. The dose of IFN-α was gradually reduced to 3 MU daily. In May 1999 she collapsed and was admitted to hospital. On admission, she complained of paraesthesiae in her hands and feet. Weight loss of 23 kg was documented and carpopedal spasm was witnessed by the admitting doctor. She had bilateral ankle oedema and stool examination revealed steatorrhoea. At the time of admission she was found to have macrocytic anaemia, haemoglobin 10.3 g/dl, mean corpuscular volume 103.3 fl, white cell count 5.2×109/l, and platelets 376×109/l. She was hypokalaemic, hypocalcaemic, hypomagnesaemic, hypoalbuminaemic, and folate deficient. Morphological assessment of duodenal biopsy specimens showed total villous atrophy and crypt cell hyperplasia (fig 1).
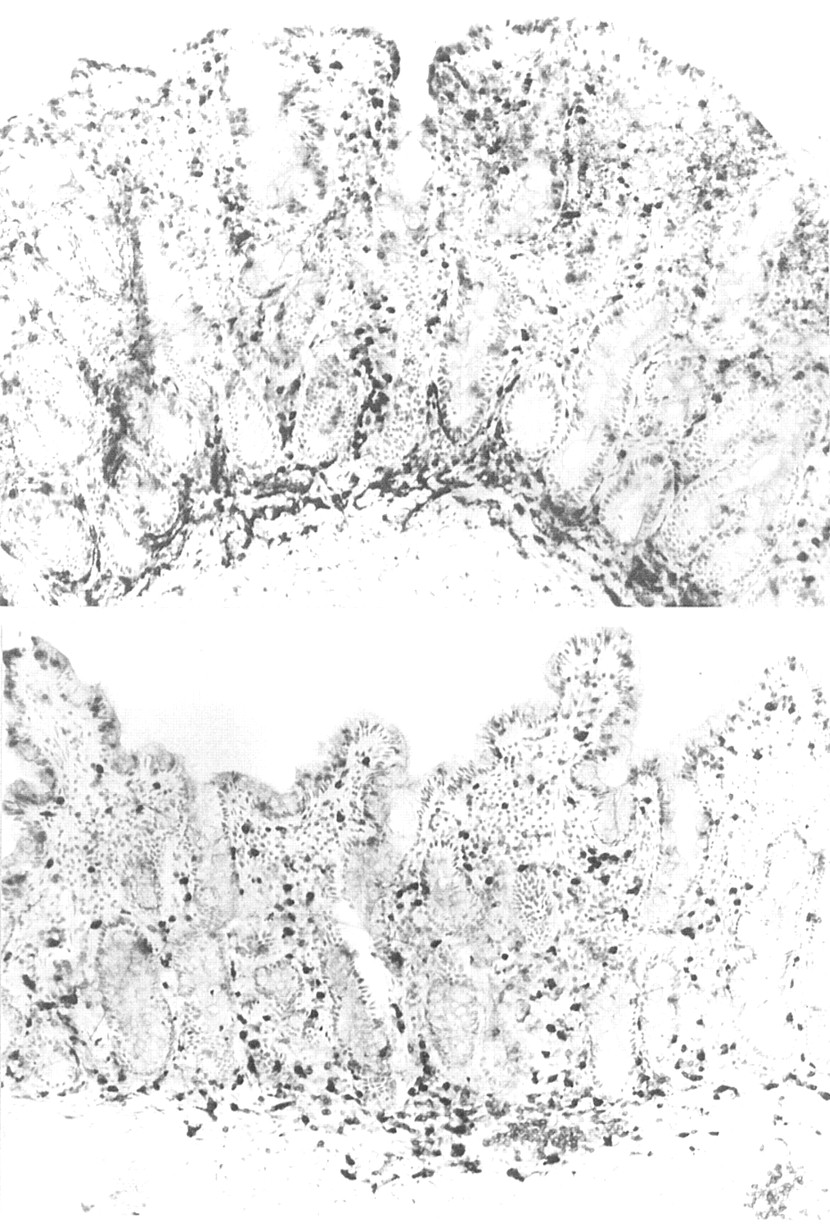
Frozen sections of duodenal biopsies taken from the patient receiving interferon α therapy before (top) and after (bottom) commencing a gluten free diet. Before the gluten free diet, the mucosa is flat, there is a dense infiltrate of intraepithelial lymphocytes (IEL), and there are a large number of non-specific inflammatory cells, as shown by the strongly positive endogenous peroxidase cells in the lamina propria. After four months of a gluten free diet, there are short villi, fewer IELs, and non-specific inflammatory cells are also markedly reduced. Immunoperoxidase with anti-CD3; original magnification ×120.
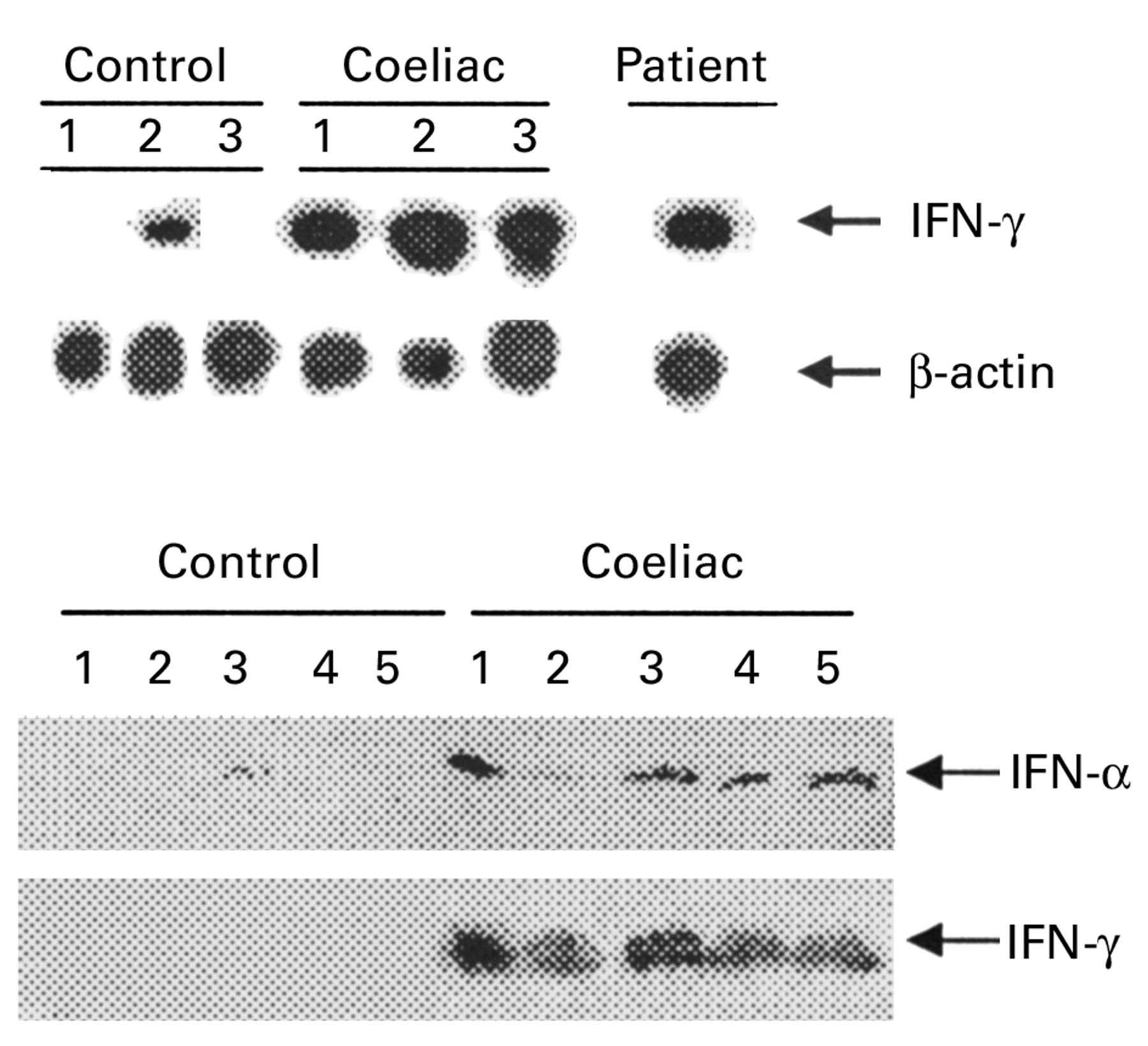
Immunostaining showed a high number of CD3+ IEL (55 IEL/100 enterocytes) (fig 1). There was no increase in epithelial γδ T cells (<1/100 epithelial cells). Colonoscopy was normal. Tissue typing revealed that the patient had the HLA DQ2 haplotype. AGA and EMA were positive. Analysis of duodenal mucosal RNA showed enhanced transcripts for IFN-γ similar to that detected in patients with proven CD (fig 2, top). Cessation of IFN-α therapy resulted in substantial clinical improvement, although loose stools continued and there was no weight gain. A strict gluten free diet was commenced and all abdominal symptoms resolved. Her weight increased to 75 kg in December 1999 with normal laboratory parameters, including AGA and EMA. Repeat duodenal biopsies showed patchy improvement with appearance of short villi and a decrease in CD3+ IEL (20.7/100 enterocytes).

{kind=link}
{kind=link}
(Top) Southern blot analysis of transcripts for interferon (IFN) γ and β-actin in duodenal mucosal tissue homogenates from three normal controls, three untreated patients with coeliac disease (CD), and the patient with chronic myeloid leukaemia receiving IFN-α treatment before commencing a gluten free diet. A total of 3 or 1.5 μl of cDNA was amplified using specific primers for IFN-γ (23 cycles) or β-actin (19 cycles), respectively. RT-PCR products were separated on agarose gel, blotted, and hybridised with specific probes for IFN-γ or β-actin. (Bottom) Western blot analysis of IFN-α and IFN-γ protein expression in mucosal samples taken from the distal duodenum of five normal controls and five patients with untreated CD. IFN-α protein was detected in mucosal samples of all CD patients and in one (lane 3) normal control. After stripping, the blot was incubated with an anti-IFN-γ antibody. A protein of approximately 19 kDa was detected only in mucosal samples of CD patients. One of two representative experiments is shown.
IFN-α IS EXPRESSED IN CD MUCOSAL TISSUE
To investigate if IFN-α is expressed in CD tissue, western blot analysis was performed using total protein extracted from duodenal mucosal samples of CD patients and controls. In all CD patients, anti-IFN-α antibody detected a protein with a molecular size of 19 kDa, comigrating with recombinant human IFN-α on sodium dodecyl sulphate-polyacrylamide gel electrophoresis (fig 2, bottom). IFN-α was only seen at very low levels in the mucosa of one control subject. In the gut mucosa of patients with CD, IFN-γ was consistently associated with IFN-α expression (fig 2, bottom). No transcripts for IL-12/P40 were detected in mucosal samples of either CD patients or controls (not shown).
Discussion
In this study, we first reported a case of CD-like enteropathy, responsive to gluten elimination, in a patient receiving IFN-α treatment for leukaemia. As the patient was also positive for AGA and EMA, we believe it is fair to conclude that the patient had CD. Secondly, we have shown for the first time the presence of IFN-α in mucosal samples from untreated CD patients but not in controls. Collectively, these data are consistent with previous reports demonstrating that IFN-α can activate autoreactive cells and promote the development of T cell responses to autoantigens,9-11but this is the first time that IFN-α has been implicated in the development of a Th1 response to non-self antigens. Indeed, we found no association between CD and IFN-α therapy in the literature. In a recently published letter, the occurrence of two cases of CD during treatment with IFN-α was indicated but there were no details on the patients.13
We cannot exclude the possibility that the patient had latent or silent CD before IFN-α treatment. We were unable to locate stored sera obtained prior to IFN-α therapy for measurement of EMA which would have helped to answer this question. None the less, the fact that the patient developed overt significant CD after IFN-α treatment indicates that the treatment either precipitated the lesion de novo or exacerbated a mild clinically silent lesion.
Although orally administered IFN-α has been reported to be beneficial in ameliorating immune diseases, there is a large body of evidence indicating the tendency of IFN-α to induce autoimmunity.9-11 ,14 ,15 The main reason for this is thought to be via induction of increased levels of costimulatory molecules such as B7.2 and ICAM-1 on APC.9 ,16 In normal circumstances where low levels of self antigen are presented on the MHC molecules of APC, a low affinity T cell receptor interaction with peptide/MHC coupled with low expression of costimulatory molecules on APC is insufficient to trigger T cell activation. However, overexpression of costimulatory molecules leads to enhanced costimulation and T cell activation against self peptides.8 The reason this patient received IFN-α was because of its ability to induce apoptosis of myeloid leukaemia cells, hence its therapeutic efficacy.9 However, IFN-α has opposite effects on normal T cells in that it can prevent apoptosis induced by IL-2 deprivation of Th1 and Th2 clones in vitro.17 Similarly, when T blasts are cultured with IFN-α, the cells leave the cell cycle and enter a resting state but do not die.18 Thus in the context of this patient, the effects of IFN-α are likely to be related to its ability to prevent T cell apoptosis and to increase costimulatory molecules on APC.
In the gut there is a delicate balance between the need to recognise antigens of pathogenic micro-organisms and the need to prevent unwanted immune responses to foods or the normal flora. The site at which this occurs is in the organised lymphoid tissue of Peyer's patches.19 We have recently demonstrated that T cells in human Peyer's patches are sensitised to dietary proteins but that this is predominantly a Th1-type pathway due to the high local expression of IL-12.20 This response was demonstrated to β-lactoglobulin of cow's milk but there is no a priori reason that the same response should not occur to wheat proteins. Following sensitisation in Peyer's patches, CD4 cells leave via the lymphatics, enter the blood, and migrate back to the lamina propria.19These cells are actively secreting IFN-γ. They do not cause disease however because they rapidly undergo apoptosis. They all express Fas, a proportion express FasL, and they express low levels of the Bcl2 survival gene.21 In addition, T cells in the lamina propria are actively downregulated. There is low level expression of B7.1 and 2 on lamina propria APC and there is high local concentrations of the immunosuppressive cytokine IL-10 and the non-specific immunosuppressive prostaglandin E2.19 ,22 It is noteworthy that in an animal model, inhibition of prostaglandin E2 can result in T cell mediated responses to food proteins and crypt hyperplasia.22
Although there is no direct evidence, data produced in other systems suggest that a possible reason why this patient developed CD is because IFN-α prevented apoptosis of lamina propria T cells and increased local expression of accessory molecules in the lamina propria. Gluten reactive CD4+ T cells arriving from Peyer's patches could therefore recognise low amounts of dietary gluten presented on lamina propria APC and be triggered because of the high expression of costimulatory molecules. Another potential contributor to the development of excess Th1 responses to gluten in the lamina propria could be due to the fact that IFN-α, after binding to its receptors on T cells, activates transcription factors (for example, STAT1 and STAT4) which then bind to the IFN-γ promoter.9Consistent with this we showed that activation of T cells in the lamina propria of fetal gut explants with IFN-α resulted in marked expression of Th1 cytokines (manuscript in preparation). Finally, it has been demonstrated recently that signalling cross talk occurs between IFN-α/β and IFN-γ, and that IFN-γ mediated cellular events are dependent on IFN-α/β signalling.23 When inflammation is initiated, local fibroblasts and macrophages produce sufficient IFN-α to maintain Th1 responses,9 which is why the patient's gastrointestinal symptoms did not improve on cessation of IFN-α therapy. This may also be the explanation for the elevated concentrations of IFN-α in untreated CD mucosa.
We do not yet know what induces IFN-α and how IFN-α production is regulated in CD. IFN-α is produced by APC, mostly in response to viruses.9 Although IFN-α synthesis is normally a transient phenomenon, abnormal IFN-α expression has been documented in humans in the absence of clinically evident infections.9 ,18 It is therefore conceivable that low grade viral infections in the gut may elevate local concentrations of IFN-α and in a genetically susceptible individual (for example, HLA-DQ2 positive) may be an important determinant in generating the persistent Th1 gluten reactive cells in the lamina propria which cause the disease.
Taken together, our data are consistent with studies showing the ability of IFN-α to promote the development or exacerbate Th1 mediated diseases.24 ,25 On the other hand, IFN-α has proved to have beneficial rather than detrimental effects in patients with Crohn's disease, an intestinal inflammatory disorder characterised by an IL-12 dependent Th1 response.24 ,26The reason why IFN-α differently modulates immune responses in the gut is unclear. However, it is known that the inflammation ongoing in the intestine of patients with Crohn's disease, but not CD, is associated with an abnormal response to antigens of the intestinal flora.27 Interestingly, type I IFN has been reported to inhibit the synthesis of inflammatory molecules, including IL-12, by bacteria stimulated APCs,28 and thus this mechanism may be responsible for the beneficial effect of type I IFN in Crohn's disease.
Acknowledgments
This work was supported by a European Union grant (ERBFMRXCT9)
Abbreviations used in this paper
- CD
- coeliac disease
- EMA
- antiendomysial antibodies
- AGA
- antigliadin antibodies
- Th1,T helper cell type 1
- IFN, interferon
- IL
- interleukin
- IEL
- intraepithelial lymphocyte
- APC
- antigen presenting cell
References
Linked Articles
- Correction