Article Text

Abstract
BACKGROUND Inflammatory bowel disease (IBD) is associated with changes in colonic motility which may contribute to the pain and diarrhoea associated with exacerbations of this disease. These changes may be mediated by prostaglandins which are increased in this condition. Increased expression of the inducible isoform of cyclo-oxygenase (COX-2) has been found in active IBD although its cellular distribution remains uncertain.
AIMS To evaluate the cellular distribution of COX-2 in active IBD.
PATIENTS AND METHODS Using reverse transcription-polymerase chain reaction, in situ hybridisation, and immunohistochemistry, COX-2 expression was evaluated in 12 colectomy specimens from patients with active ulcerative colitis (UC), and six specimens from patients with Crohn's colitis that had failed medical therapy. Histologically normal colon was obtained from 12 patients having resection for colorectal neoplasia and evaluated as above, acting as control specimens.
RESULTS All specimens expressed COX-2 mRNA, with some 6–8-fold increase in inflamed tissues on densitometric analysis (both UC and Crohn's) compared with controls. In situ hybridisation localised this mRNA to myenteric neural cells, surrounding smooth muscle cells, and inflammatory cells of the lamina propria in the IBD specimens, with some weaker labelling seen in the epithelium. No COX-2 labelling was seen in normal tissues. Immunohistochemistry confirmed these sites of COX-2 expression in all inflamed specimens, with absence of immunoreactivity in control tissues.
CONCLUSIONS These findings provide the first evidence of COX-2 expression in neural cells of the myenteric plexus in active IBD which, via increased prostaglandin synthesis at this site, may contribute to the dysmotilty seen in this condition.
- inducible cyclo-oxygenase
- prostaglandins
- myenteric plexus
- inflammatory bowel disease
Abbreviations used in this paper
- IBD
- inflammatory bowel disease
- PGE2
- prostaglandin E2
- COX
- cyclo-oxygenase
- RT-PCR
- reverse transcription-polymerase chain reaction
- G3PDH
- glyceraldehyde-3-phosphate dehydrogenase
- SSC
- standard saline citrate
- UC
- ulcerative colitis
- PGP
- protein G peptide
- iNOS
- inducible nitric oxide synthase
Statistics from Altmetric.com
For many years it has been appreciated that there is altered colonic motility in active inflammation of the colon.1Such alterations in colonic motor and myoelectric activities with resultant changes in transit may contribute to the pain and diarrhoea characteristic of an exacerbation of inflammatory bowel disease (IBD).2 The mechanisms of this dysmotility are uncertain but altered function of myenteric nerves and smooth muscle cells have been reported.2
Proinflammatory cytokines alter enteric nerve function which in the rat is thought to be mediated via prostaglandin production (PGE2 primarily).3 Prostanoids are known to play significant physiological and pathophysiological roles in the gastrointestinal tract; regulating water and electrolyte transport, mucus secretion, blood flow, and motility.4 There is evidence of increased synthesis of PGE2 in the rat myenteric plexus after appropriate cytokine stimulation3and also in the rectal mucosa of patients with active IBD.5-7 PGE2 has also been shown to modulate both non-adrenergic and non-cholinergic neurotransmission in the gastrointestinal tract of the rabbit.8
Further known effects of this prostanoid include stimulation of acetylcholine release from guinea pig myenteric plexus neurones with resultant changes in colonic contractility suggesting an effect via neural mechanisms.9 However, the effects of PGE2 on colonic motility in vivo are less certain. A decrease in sigmoid contractility occurs in humans during intravenous infusion of PGE2,10 whereas stimulation of the distal colon occurs in the rabbit after a similar infusion,11 suggesting that the effects of prostaglandins on the control of colonic motility may be species specific.
Prostaglandin receptors have been reported on both enteric neural and smooth muscle cells of the guinea pig ileum,12 ,13 but as yet there are no known reports of similar receptor expression in the human gastrointestinal tract.
There is increased expression of the inducible isoform of cyclo-oxygenase (COX-2) in both an animal model of colitis and in human IBD14-16 but with conflicting data on cellular localisation. In the animal model, COX-2 was primarily expressed in the subepithelium14 whereas epithelial expression was shown in human colitis15 with no study to date reporting COX-2 expression in neural cells of the colonic myenteric plexus. Therefore, we examined expression and localisation of this inducible isoform in full thickness surgical specimens, taken from patients with refractory IBD.
Materials and methods
PATIENTS AND SPECIMENS
Histologically normal tissue was obtained from colectomy specimens from patients with colorectal neoplasia from a site distant from the lesion (n=12) and surgical specimens were obtained from a similar site in patients undergoing surgery for refractory ulcerative (n=12) and Crohn's (n=6) colitis. Tissue was immediately frozen and stored at −70°C and post fixed as required. Characteristics of the patient groups are shown in table 1.
Characteristics of patient groups
REVERSE TRANSCRIPTION-POLYMERASE CHAIN REACTION (RT-PCR)
Tissue samples were collected as discussed, and immediately frozen in liquid nitrogen. Total RNA was purified from 200 mg of tissue according to the method of Chomczynski and Sacchi.17 This entailed homogenisation in guanidine thiocyanate solution, acid phenol/chloroform extraction, and isopropanol precipitation. RNA pellets were washed in 70% ethanol, dissolved in sterile water, and stored at −70°C. Aliquots of each RNA sample (5 μg) were reverse transcribed to produce single stranded cDNA with a kit (Invitrogen, USA) using random primers and avian myeloblastosis virus reverse transcriptase. Samples of cDNA (50 ng) were then analysed by PCR amplification using pairs of oligonucleotide primers specific for COX-2. Primer sequences were: 5′ gTT CCA CCC gCA gTA CAg 3′ sense, and 5′ ggA gCg ggA AgA ACT TgC 3′antisense primer, amplifying a 483 bp cDNA fragment.
Each PCR reaction contained 1× NH4 CL PCR buffer, 1.875 mM MgCl2 , 0.1 μM of each primer, 0.5 mM dNTPs, 1 μCi32P-dCTP, and 2.5 units of BioTaq DNA polymerase (Bioline, UK). Following five minutes of denaturation at 93°C, hot start reactions were initiated (annealing at 60°C for 30 seconds, extension at 72°C for 30 seconds, and denaturation at 93°C for 30 seconds) and run for up to 35 cycles. Semiquantitative PCR data were generated by kinetic analysis and comparison with levels of glyceraldehyde-3-phosphate dehydrogenase (G3PDH) mRNA. Reaction products were analysed by 6% polyacrylamide gel electrophoresis and film autoradiography using radiolabelled DNA size markers. Amplification products separated in this way were excised and eluted from the gel into 0.5 M ammonium acetate and ethanol precipitated. The identity of the product was confirmed by direct cycle sequencing using a kit (Circumvent, New England Biolabs, USA).
IN SITU HYBRIDISATION
Fresh tissue samples were fixed by immersion in 1% paraformaldehyde in phosphate buffered saline. Samples were then frozen in cryostat embedding compound for preparation of 10 μm sections on 3-aminopropyltriethoxysilane coated slides. Sense and antisense32P labelled single stranded DNA probes were generated by PCR. Riboprobes were generated using T7 RNA polymerase after making templates by PCR with an appropriate T7 promoter tagged primer. Probes were purified by phenol/chloroform extraction and ethanol precipitation and were washed six times with 70% ethanol to remove unincorporated nucleotides. The probes were dissolved in water, denatured at 90°C for one minute, cooled on ice, and serially diluted in hybridisation buffer (50% formamide, 6× standard saline citrate (SSC), 5× Denhardt's solution (containing bovine serum albumin, ficoll, and polyvinyl pyrrolidone), 100 μg/ml sheared and denatured herring sperm DNA, and 100 μg total RNA from baker's yeast.) Sections were hybridised under siliconised coverslips at 42°C overnight in humidified chambers. Coverslips were removed in 5× SSC at room temperature and racks of slides were washed at 42°C in 2× SSC, 1× SSC, and 0.5× SSC for 20 minutes in each solution. Washed slides were allowed to dry before performing film autoradiography (with Amersham Hyperfilm B-max, 24–72 hour exposure) followed by emulsion autoradiography (2–5 days with Ilford K5 liquid emulsion). Silver grains were developed using Kodak D-19 developer and fixed in dilute Amfix (Champion, UK). Sections were counterstained, coverslipped, viewed, and photographed.
IMMUNOHISTOCHEMISTRY
COX-2 protein expression was examined in tissue using a standard ABC Vectastain technique. Tissue sections were post fixed in 1% paraformaldehyde. Briefly, endogenous peroxidase activity was blocked in tissue sections (10 μm sections on 3-aminopropyltriethoxysilane coated slides) using endogenous peroxidase suppressor “Immunopure” (Pierce Laboratories) for 22 minutes followed by application of normal goat serum (1:20) to block non-specific staining for 30 minutes. The primary antibody, a rabbit polyclonal raised against COX-2, was obtained from Cayman Chemical (Ann Arbor, Michigan, USA). This was applied to serial sections at a concentration 1:1000 and incubated for one hour at room temperature. Following this the second antibody was applied, a goat antirabbit biotinylated antibody (applied at 5 μg/ml, obtained from Vecta Laboratories, California, USA). This was further probed with AB Vectastain and developed with diaminobenzadine tetrahydrochloride dihydrate for 3–5 minutes, with resultant brown staining indicating sites of COX-2 immunolocalisation. Normal rabbit serum (concentration up to 2 μg/ml) was applied on serial sections as a negative control. Sections were counterstained with haematoxylin (1:20). Localisation of COX-2 was compared with that of the neural marker protein G peptide (PGP9.5) and COX-1 (the constitutive isoform of cyclo-oxygenase) using appropriate antibodies against these proteins.
Results
Using RT-PCR all specimens, normal and diseased, were shown to express COX-2 mRNA. Kinetic PCR analysis revealed increased COX-2 expression (up to eightfold on densitometry) in 12/12 ulcerative colitis (UC) and 5/6 Crohn's specimens compared with normals when standardised to the level of G3PDH expression (fig 1). In situ hybridisation localised COX-2 mRNA to the myenteric plexus (neural cells and smooth muscle) in all UC specimens (12/12) evaluated and in 5/6 of the Crohn's specimens, with labelling also seen in inflammatory cells of the lamina propria in all of these inflamed tissues. No COX-2 labelling was seen in normal controls (fig 2). Immunohistochemistry localised COX-2 protein to myenteric neural and smooth muscle cells in all UC and in 5/6 Crohn's specimens. PGP was coexpressed at these sites confirming neural cell expression of COX-2 in the inflamed tissues. There was also COX-2 immunoreactivity in the epithelium of 7/12 UC and 4/6 Crohn's specimens with absence of COX-2 immunoreactivity at any site in normal tissue (fig3).

Example of kinetic polymerase chain reaction (PCR) analysis of COX-2 expression in normal (1) and ulcerative colitis (UC) (2) tissue. Densitometric analysis revealed greater expression (up to eightfold) of COX-2 product in 12/12 UC and 5/6 Crohn's specimens compared with normal tissue when standardised with respect to glyceraldehyde-3-phosphate dehydrogenase (G3PDH) levels. PCR cycle lengths: G3PDH 30–34, COX-2 31–35. M, DNA size markers from Gibco BRL, UK.

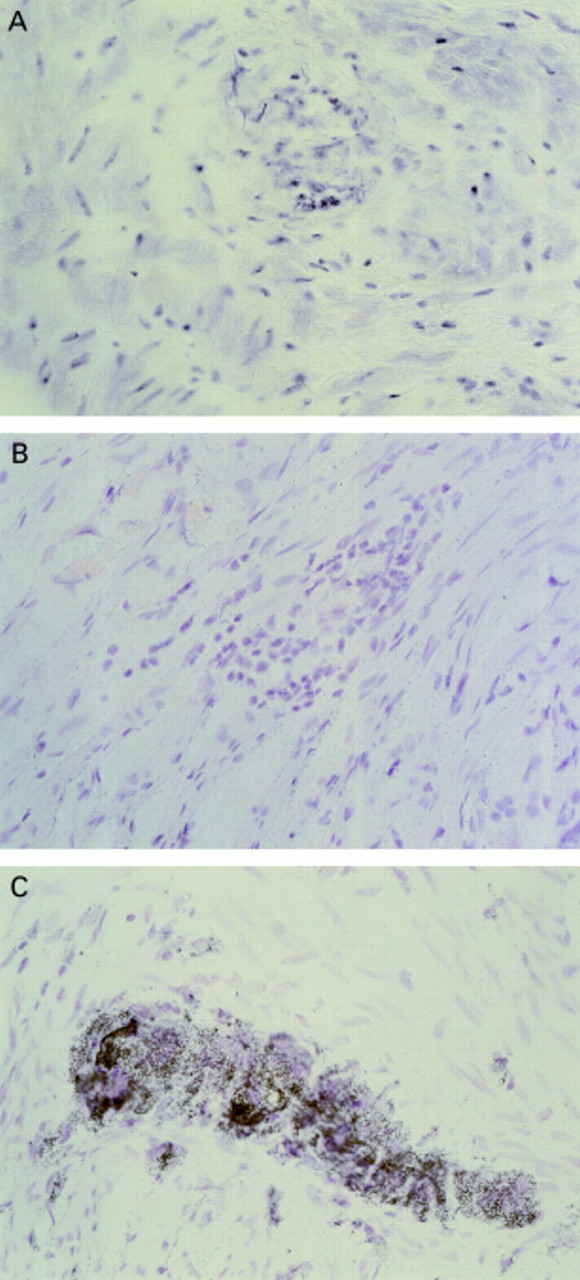
Example of COX-2 in situ hybridisation of surgical resection specimens. (A) Non-inflamed control tissue and antisense probe, with no labelling seen. (B) Diseased tissue (ulcerative colitis (UC) specimen) and sense probe—no labelling. (C) Diseased tissue (UC) and antisense probe with intense labelling of neural cells of the myenteric plexus. Similar results were obtained irrespective of the mode of inflammation—that is, in both UC and Crohn's specimens COX-2 mRNA was localised to the same cell types.
{kind=link}
{kind=link}
{kind=link}
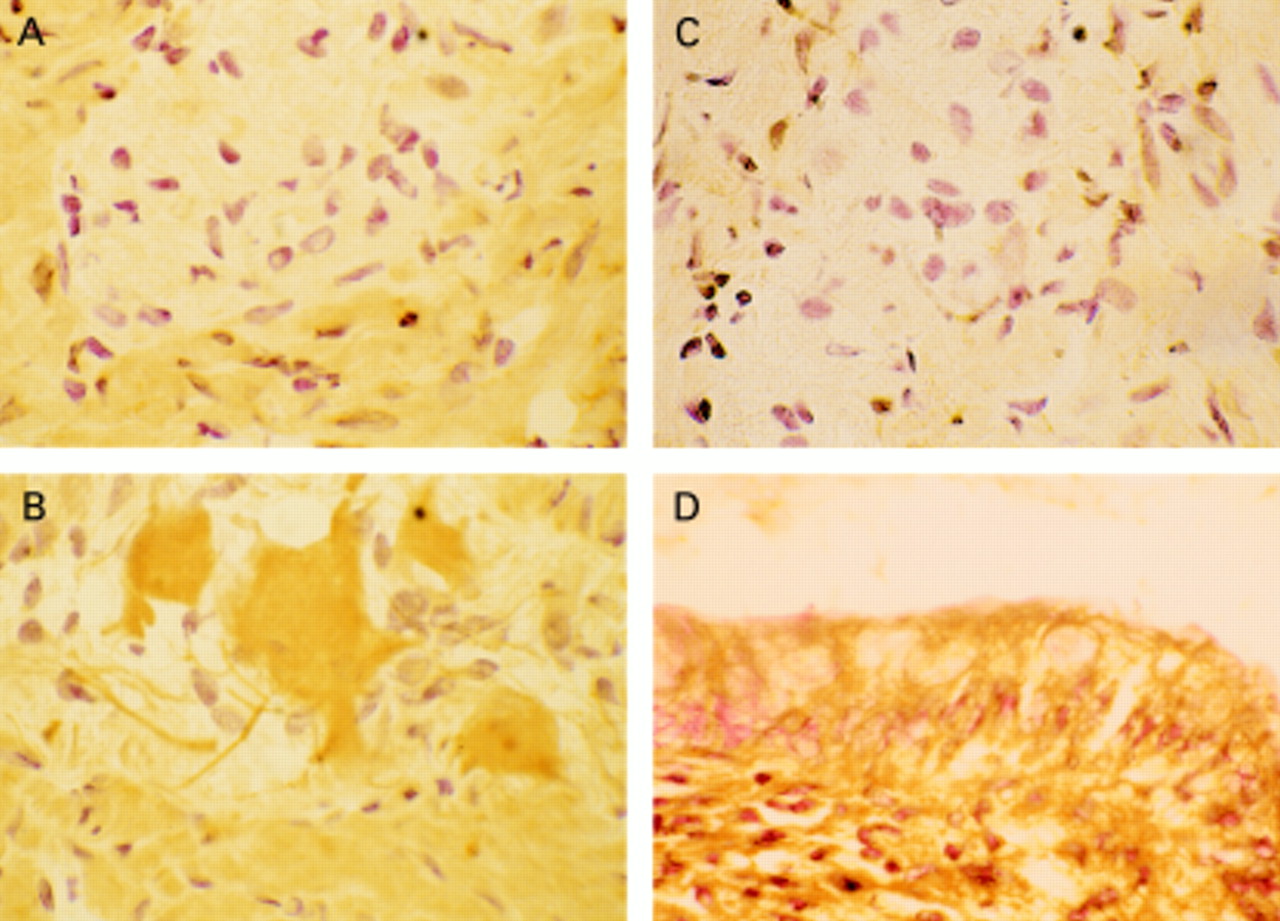
Example of immunolocalisation of COX-2. (A) Non-immune (control) serum-diseased tissue (ulcerative colitis (UC)), with no staining seen. (B) COX-2 labelling (brown stain) of neural cells of the myenteric plexus in diseased tissue (UC). (C) Absence of COX-2 labelling in myenteric plexus of normal tissue. (D) Labelling of epithelial cells with COX-2 in diseased tissue (UC).
Discussion
Our data confirm previous studies suggesting increased COX-2 expression in active IBD,14-16 with labelling of the colonic epithelium and inflammatory cells of the lamina propria. However, we have also shown expression of this enzyme in neural cells of the myenteric plexus in active colonic inflammation, an as yet unpublished finding.15 Using antibodies to both neural cells (PGP9.5) and macrophages (anti-CD68, data not shown) we were able to show specific neural cell expression of COX-2 in the human myenteric plexus and not in activated macrophages at this site, as suggested in a previous report.3 The subepithelial expression of COX-2 is consistent with the findings in TNBS induced colitis in rats14 which with transmural inflammation is histologically more similar to Crohn's colitis than UC. Therefore, we were not surprised to find COX-2 production in the myenteric plexus in Crohn's colitis, although no neural cell labelling was seen in the TNBS animal model. The finding of COX-2 expression at this site in the UC specimens was less expected as UC is felt to be, in the main, a mucosal disease.
In certain cell systems a recognised stimulus for COX-2 expression is mechanical stress which would have occurred during the manipulation and resection of the colonic specimens.18 However, the absence of any COX-2 expression in both smooth muscle cells and neural tissue in the normal specimens (using localisation techniques) would suggest that such physical effects play no role in the expression of COX-2 in our specimens, as all tissue examined was obtained in a similar manner (that is, resection at laparotomy).
In studies of patients with chronic ulcerative colitis there is reduced colonic contractility,19 reduction in basal colonic intraluminal pressure,20 and a marked reduction in postprandial motility.21 In both human and animal models of colitis there appears to be a relationship between altered colonic motility and abnormal local release of various inflammatory mediators of which prostaglandins such as PGE2 are of considerable importance.22 ,23
Toxic megacolon is a severe and life threatening consequence of the dysmotility of acute IBD. Mucosal nitric oxide (NO•) is increased in active IBD24-26 and a recent report evaluating expression of the inducible isoform of nitric oxide synthase (iNOS) in UC suggested increased production of this inflammatory enzyme in the muscle layers of the colon and implicated the resultant nitric oxide as the mediator of the reduced contractility and consequent toxic megacolon.27 None of our specimens was taken from patients with toxic dilatation of the colon which may explain the absence of iNOS expression in the muscle layers of these tissues in our study (data not shown). The role of COX-2 at this site in toxic megacolon is still poorly understood. However, with COX-2 expression in both smooth muscle and neural cells of the myenteric plexus in active colitis, it is plausible that the resultant prostanoids from COX-2 enzyme activity at this site might be of importance in the pathogenesis of toxic megacolon.
It has been suggested that selective inhibition of COX-2 is beneficial in states of inflammation.28 This may not be the case in acute colitis however as such inhibition in an animal model of colitis increased mortality,14 with no similar human study to date. Therefore, although there is increased production of COX-2 in active IBD it is not known whether using selective COX-2 inhibitors would be beneficial, although this remains a possibility, specifically with regards to toxic colonic dilatation.
In summary, we have shown a new finding of COX-2 expression in neural cells of the myenteric plexus during active IBD, and suggest that such enzyme expression at this site may link colonic inflammation, prostaglandins, and colonic dysmotilty.
Abbreviations used in this paper
- IBD
- inflammatory bowel disease
- PGE2
- prostaglandin E2
- COX
- cyclo-oxygenase
- RT-PCR
- reverse transcription-polymerase chain reaction
- G3PDH
- glyceraldehyde-3-phosphate dehydrogenase
- SSC
- standard saline citrate
- UC
- ulcerative colitis
- PGP
- protein G peptide
- iNOS
- inducible nitric oxide synthase