Article Text

Abstract
Familial adenomatous polyposis (FAP) is characterised by the presence of profuse colonic carpeting of adenomas throughout the entire colon and rectum. The genetic basis of FAP has been shown to be primarily associated with germline mutations in theAPC gene. Notwithstanding, several reports have been published indicating that there is genetic heterogeneity in FAP and that the most likely explanation is the existence of another gene. In this report we further delineate the genotype/phenotype correlation in families that harbour germline mutations in theAPC gene and identify some previously unreported changes in the APC gene which predispose to an attenuated disease phenotype. From 53 index patients diagnosed with either FAP or attenuated FAP, 27 harboured changes in the APC gene. The remaining 26 patients were further subgrouped according to their colonic phenotype. There were nine patients with a mixed hyperplastic/adenomatous colonic phenotype and there were 17 patients with an adenomatous colonic phenotype. Evaluation of the disease characteristics of these patients and their families is presented which may aid in the identification of new genes associated with colonic polyposis.
- familial adenomatous polyposis
- genetics
- hyperplastic polyposis
- attenuated adenomatous polyposis coli
- hereditary non-polyposis colorectal cancer
Abbreviations used in this paper
- FAP
- familial adenomatous polyposis
- APC
- adenomatous polyposis coli
- AAPC
- attenuated adenomatous polyposis coli
- CHRPE
- congenital hypertrophy of the retinal pigment epithelium
- SSCP
- single strand conformation polymorphism
- DGGE
- denaturing gradient gel electrophoresis
- PTT
- protein truncation test
- ARMS
- amplification refractory mutation system
- HNPCC
- hereditary non-polyposis colorectal cancer
- PCR
- polymerase chain reaction
Statistics from Altmetric.com
- familial adenomatous polyposis
- genetics
- hyperplastic polyposis
- attenuated adenomatous polyposis coli
- hereditary non-polyposis colorectal cancer
Familial adenomatous polyposis (FAP) is characterised by the development of hundreds to thousands of adenomas throughout the entire colon and rectum, which if not removed will almost certainly result in the development of colorectal cancer.1 Variance in disease expression has also been described and is typified by fewer colonic and rectal adenomas (usually less than 100), later age of onset, and a reduced likelihood of colorectal cancer development. Variant forms of FAP are known as attenuated adenomatous polyposis (AAPC) or the flat adenoma syndrome.2 ,3 The incidence of FAP in the population is approximately 1:7000 according to the Danish Polyposis Registry and it affects both sexes equally.
The genetic basis of FAP was first localised in 1986 to chromosome 5q and the region localised to 5q21 in 1987.4 ,5 In 1991 a gene for FAP was identified and called theAPC gene.6-9 APC mRNA is 8531 bp in size and codes for a protein of approximately 310 kDa. The APC protein is multifunctional and its primary role appears to be linked to the breakdown of β-catenin via the ubiquitin degradation pathway.10Several other proteins not associated with that pathway have been shown to bind to the APC protein, suggesting a diversity of function.11 ,12
Identification of mutations within the APCgene has revealed the existence of genotype/phenotype correlations between the site of mutation and disease expression. Mutations restricted to 5′ of codon 169, exon 9, and 3′ of codon 1403 in exon 15 have been associated with an increased likelihood of a mild colonic phenotype.3 ,13-15 In addition, mutations beyond codon 1403 are associated with a much greater probability of extracolonic disease.14-17 Germline mutations between exon 9 and approximately codon 1400 have been correlated with the presence of congenital hypertrophy of the retinal pigment epithelium (CHRPE). Outside of this region, CHRPE also occurs but there is a tendency for these mutation carriers not to express this sign.18Finally, a common variant of the APC gene has been identified in the Jewish population, leading to lower penetrance than most APC mutations. This variant is an amino acid change at codon 1307 where a lysine substitutes for a isoleucine residue and is associated with an attenuated disease phenotype. This alteration of theAPC gene is the first genetic change that has been identified which cannot be unequivocally assigned as causative.19
Several screening strategies have been developed to identify mutations in the APC gene. They include single strand conformation polymorphism analysis (SSCP), heteroduplex analysis, denaturing gradient gel electrophoresis (DGGE), the protein truncation test (PTT), and direct sequencing. Despite these various mutation detection strategies, 20–50% of families do not appear to have changes in the APC gene.20 It is unlikely that the screening methods employed forAPC gene mutation detection are incapable of detecting such a high proportion of cases. Linkage analysis in one series clearly identified the presence of genetic heterogeneity21 which may explain some of these findings. More recently, Heinimann et al identified phenotypic differences in FAP patients who did not harbour germline mutations in the APC gene. Typically,APC mutation negative patients were older than their APC mutation positive counterparts and presented with fewer extracolonic symptoms. Colonic disease in the APC mutation negative patients tended to be less severe as they usually presented with fewer than 100 colonic adenomas compared with theAPC mutation positive patients who presented with more than 100 colonic adenomas.20
In this study we have analysed 53 unrelated patients who fulfilled the clinical criteria of FAP or attenuated FAP. Mutation analysis was performed using DGGE for exons 1–14 and the PTT assay for exon 15. All polymorphisms and truncated proteins were subjected to DNA sequencing for confirmation and characterisation. All families that did not harbour any obvious change in the APC gene were further studied to determine if they harboured the I1307KAPC polymorphism. Phenotypic characteristics were analysed in a total of 194 affected individuals and compared according to their APC mutation status.
Patients and methods
A group of 53 unrelated patients diagnosed with colonic polyposis at colonoscopy or colectomy were examined for mutations in theAPC gene. All probands gave informed consent for genetic testing. Of the 53 patients, 12 presented with colonic and extracolonic disease, four were described as having attenuated disease, and there were 14 patients who did not have a family history of disease. The remaining 23 patients only presented with colonic polyposis but did have strong family histories of colorectal cancer consistent with an autosomal dominant inheritance pattern. The diagnostic criteria for FAP were the presence of more than 100 adenomas throughout the colon and rectum. Included in the study were patients who satisfied the diagnosis of attenuated FAP. This included patients with a family history of disease that was associated with multiple colonic adenomas (>20) at a relatively young age.
Genomic DNA was isolated from Na2EDTA blood according to the method described by Miller and colleagues.22 Briefly, blood samples were mixed with EL buffer (155 mM NH4Cl, 10 mM KHCO3, 1 mM Na2EDTA, pH 7.4) and left on ice for 15 minutes. The lysate was centrifuged, washed twice with EL buffer, and the intact lymphocyte pellet resuspended in NL buffer (2 ml; 100 mM Tris HCl, pH 8.0, 0.1 M NaCl, 2 mM Na2EDTA, 1% SDS, and 1 mg/ml proteinase K), and incubated overnight at 55°C. The next day, 333 μl/ml of 6 M NaCl was added, mixed, and centrifuged to remove cellular proteins. The supernatant containing the DNA was placed in a fresh tube and the DNA precipitated with ethanol. The resulting DNA pellet was washed with 70% ethanol, dried briefly, and dissolved in TE buffer (10 mM Tris HCl, pH 8.0, 1 mM Na2EDTA) to a concentration of 500 ng/μl.
DENATURING GRADIENT GEL ELECTROPHORESIS (DGGE) AND AMPLIFICATION REFRACTORY MUTATION SYSTEM (ARMS) ANALYSIS
Screening of the APC gene was performed in two stages. Exons 1–14 were screened for mutations using DGGE and exon 15 by the PTT assay. The I1307K polymorphism was screened using the ARMS technique.19 Briefly, for DGGE, amplification of exons 1–14 was performed using primer pairs similar to those previously described7 except that the forward primer of each pair included a GC clamp. The polymerase chain reaction (PCR) was used to amplify genomic DNA. Amplification was performed according to the method of Fodde and colleagues.23 In summary, a 25 μl PCR mixture contained 100 ng of genomic DNA, 0.8 μM of each primer, 200.0 μM of each dNTP, 3 mM MgCl2, 1.0 U Taq DNA polymerase, and 1× reaction buffer (10 mM/l Tris HCl, pH 7.5, 50 mM/l KCl, 0.2 mg/ml BSA). The reaction parameters were one cycle of 95°C for one minute followed by 35 cycles of 95°C for one minute, 58°C for 1.5 minutes, and 72°C for two minutes. PCR products were subjected to DGGE using gradients ranging from 10% to 80% urea in 10% polyacrylamide (29:1) and visualised by ethidium bromide staining. A permanent record was made by photography. The ARMS technique was similar to that previously described24 without modification.
PROTEIN TRUNCATION TEST (PTT)
The PTT was performed essentially as described by Powell and colleagues.25 Exon 15 of theAPC gene was divided into four overlapping segments containing codons 686–1217, 1099–1693, 1555–2256, and 2131–2843. The primers used for amplification were designed to introduce a T7 promoter sequence for the initiation of transcription by T7 RNA polymerase, as well as a Kozak consensus sequence and ATG codon for initiation of translation. Similar PCR parameters were used as for DGGE analysis. PCR products were used directly as templates in 10 μl coupled transcription-translation assays (Promega, Madison, Wisconsin, USA) using 30 μCi of [35S] methionine (Amersham, Bucks, UK). Samples were incubated for one hour at 30°C, diluted in sample buffer, heated for five minutes at 95°C, and analysed on a 4–20% gradient SDS/polyacrylamide gel (Biorad). Protein was visualised by autoradiography.
DNA SEQUENCING
New DGGE polymorphisms, prematurely truncated proteins identified by the PTT, and the overlap regions of exon 15 were subject to DNA sequencing. Dideoxy sequencing of the PCR products was performed using a BIGDYE dideoxy sequencing ready reaction kit and analysed on an ABI 310 automated sequencer (PE Foster City, California, USA).
STATISTICAL ANALYSIS
Statistical differences between different groups of patients were compared using a Student's t test.
Results
All patients entered into this study had a clinical diagnosis of adenomatous (44 probands) or adenomatous and hyperplastic polyposis (nine probands). Of the 53 patients, 27 were found to harbour causative germline mutations in the APC gene and 26 patients were not found to harbour any change. The 26 patients that did not have a discernable causative germlineAPC mutation were further analysed for the I1307K mutation: none was found to harbour this change.
APC GENE POSITIVE FAMILIES
The 27 mutations identified were spread across theAPC gene with the majority clustered more towards its 5′ end (see fig 1). All but seven mutations were in exon 15 and four of these were within regions that have previously been associated with an attenuated FAP phenotype. There were three families harbouring a common mutation at codon 1061 and two unrelated families shared an identical base insertion at position 4330, which resulted in a stop codon at amino acid 1454. The common mutation at codon 1309 was not identified in this series. Most mutations were either small insertions or deletions, which gave rise to a truncated protein (see table 1 for a summary of mutations). Of particular note was the finding that four mutations that resulted in a predicted protein truncation were not identified by the PTT but were found on DNA sequencing of the overlap regions of exon 15 (see fig 1). In all patients who had a family history of disease, additional family members were tested for the identified mutation.
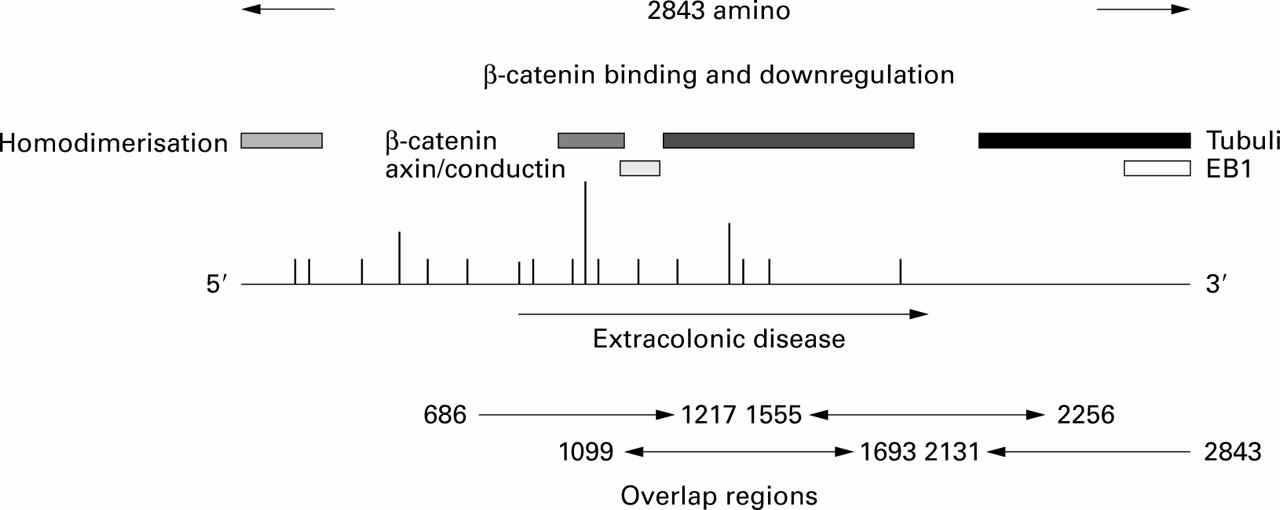
Schematic diagram of the APC gene indicating functional regions, mutation sites, and overlap regions of analysis. Proteins (including the homodimerisation region) described above the gene bind at their respective positions to APC. Vertical lines above the gene represent causative changes. Repeated mutations are indicated by the height of the lines.
Clinical characteristics of the 53 index patients tested for APC germline mutations.
Two mutations were unusual. One mutation was a 17 bp duplication of the sequence between nucleotides 5772 and 5790 inserted at position 5790, which resulted in a premature stop codon at amino acid 1969. The predicted protein was identical to the APC protein until codon 1930, thereafter the remaining 39 amino acids coded for a nonsense C terminal peptide. Patients harbouring this mutation tended to present with relatively sparse colonic polyposis and had a predilection for disease outside of the gastrointestinal tract (osteomas and desmoid disease).
The other interesting mutation was a 14 bp deletion beginning at nucleotide position 1005 through to 1019. This mutation is in exon 9 and is associated with an attenuated form of the disease. Colonic polyposis tended to be right sided with sparse presentation in the descending colon. An interesting feature described in two patients was the presence of mixed hyperplastic adenomatous polyposis. Extracolonic disease was not generally a feature in this family, but several patients presented with small cystic hamartomatous fundic gland polyps. Disease development tended to be at older ages compared with typical FAP families (34.5 (range 21–51) v 26.5 (range 11–65)) but this was not statistically significant, which is probably a reflection of the sample population.
A comparison between the mutations occurring within regions of theAPC gene previously identified as being associated with an attenuated phenotype revealed different disease characteristics compared with those families harbouring mutations associated with a typical FAP phenotype. The average age of diagnosis of the attenuated FAP patients was significantly different from typical FAP patients (p=0.019), being 42 years (range 21–73) compared with 26.5 years (range 11–65). In the attenuated FAP families, there were no reports of disease occurring at other sites apart from the gastrointestinal tract whereas 12 FAP families were associated with recognised extracolonic manifestations.
APC MUTATION NEGATIVE FAMILIES
To determine if there were specific features of the polyposis families that did not appear to harbour APCgene mutations, a comparison between age of disease onset and other clinical features was made. The average age of diagnosis forAPC mutation positive families was 28.5 years (range 11–73) with an unusual gender difference with females representing 61.1% and males 38.9% of patients. The mutation negative families were diagnosed at an average age of 41.4 years (range 8–81) and there were no sex differences. The difference in age at diagnosis between mutation positive and mutation negative families was found to be statistically significant (p=0.0004).
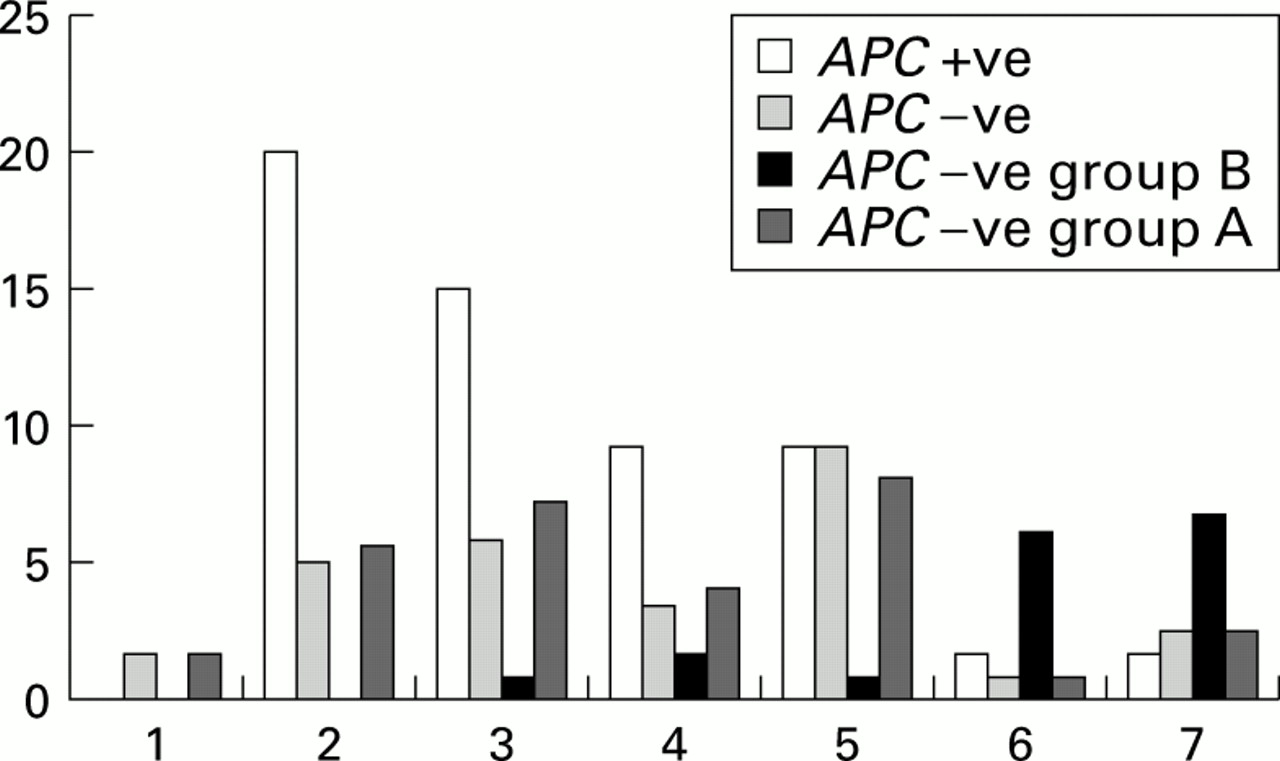
Within the mutation negative families two subgroups could be differentiated: an adenomatous polyposis group (group A) and a mixed adenomatous hyperplastic polyposis group (group B). The average age for group A was 32.7 years (range 8–72) with a sex distribution of 56.3% females and 43.7% males and for group B (excluding theAPC mutation positive family reported with a mixed colonic phenotype) 54.4 (range 11–81) and a sex distribution of 40% females and 60% males. This result was found to be highly statistically significant (p=0.00003). In fig 2 the age distribution at diagnosis is shown for group A, group B, and groups A and B compared with APC mutation positive families. The majority of mutation positive cases are diagnosed within the first three decades of life whereas the majority of patients with a mixed adenomatous hyperplastic phenotype are diagnosed in their sixth and seventh decades of life. Group A families tended to be diagnosed at an intermediate age compared with the APCmutation positive group and group B. Other disease characteristics were examined to further refine any similarities and differences between the three groups (APC gene mutation positive families, group A, and group B). The major clinical features observed between all groups are presented in tables 2-4. Desmoid disease was identified in eight mutation positive families, in one mutation negative group A family, and in none of the group B families. Osteomas were identified in four APC mutation positive families and none of the other families. Mixed adenomatous hyperplastic polyposis was observed in eight mutation negative families and in one mutation positive family (as shown in fig 3). All hyperplastic polyps described in this study were identified within the colons of affected patients and displayed typical features with serrated glandular profiles but without nuclear hyperchromasion or atypia. The distribution of these lesions in all cases was pancolonic with no apparent sites of proclivity.

Age of diagnosis of APC mutation positive families compared with mutation negative families combined and when divided into a mixed hyperplastic adenomatous colonic phenotype (group B) and an adenomatous phenotype alone (group A). Numbers on the x axis represent age ranges: 1, 1–9 years; 2, 10–19 years; 3, 20–29 years; 4, 30–39 years; 5, 40–49 years; 6, 50–59 years; and 7, >60 years.

{kind=link}
{kind=link}
{kind=link}
Presence of extracolonic disease and hyperplastic polyposis in APC mutation positive families, APC mutation negative families, and families with a mixed hyperplastic/adenomatous polyp colonic phenotype. The occurrence of desmoids in the three groups (APC mutations positive, group A, and group B) is shown in the first three categories, the presence of osteomas is shown in the next three, and mixed adenomatous/hyperplastic polyposis is shown in the last three columns
Discussion
Using a combined strategy of PTT, DGGE, and direct DNA sequencing of the overlap regions within the interval covered by the PTT, several families that otherwise would have been assigned asAPC mutation negative were found to harbour mutations. Furthermore, families in which we did not find anAPC mutation were examined for the common Jewish I1307K polymorphism by the ARMS technique.22 Using this approach the likelihood of missing mutations is very low and could not account for the number of families that were screened and found to be APC mutation negative. From our experiences we recommend this screening strategy for the analysis of the APC gene.
The mutation spectrum identified in this series of families is similar to that reported by others (reviewed in Nagase and Nakamura,26 Miyaki and colleagues,27 and Polakis28). Two unusual mutations however were identified: a 14 bp deletion and a 17 bp duplication, both of which result in an atypical polyposis phenotype. In addition to the 14 bp deletion, two more mutations were identified in exon 9, increasing the number of families reported with mutations identified within this exon.
The 14 bp deletion occurring in exon 9 has not been reported previously and is the largest genetic change that has been described for this exon. This finding adds to the small body of information about mutations in exon 9 that are associated with an attenuated disease phenotype. The differential splicing within exon 9 results in the removal of nucleotides 934–1236 from mRNA which removes 101 amino acids (residues 312–412) from the predicted APC protein.6 ,7 The resultant splice variant can be detected in normal tissue but is less abundant than the full length protein. The mutation in exon 9 described here occurs within the differential splice site and removes codons 335–340 and results in a termination codon at position 364. The presence of a mutation within this differential splice site is likely to reduce the impact of the mutant allele on disease expression and explains the mild phenotype observed in this family. Presumably, differential splicing of this change occurs which renders the mutant product less likely to be involved in disease development.29 In a recent report however it appears that intrafamilial variation30 is best explained by the presence of modifier genes but in the family reported here, the disease phenotype was remarkably consistent in that disease presentation is very predictable within this family. Notwithstanding, this family is remarkable (described in table 2); hamartomatous polyps were observed in the stomach of some individuals and one patient presented with a single hyperplastic polyp in association with adenomatous polyposis. Furthermore, in affected individuals, colonic disease was restricted to the ascending colon and there was an almost complete absence of adenomatous polyps in the descending colon or rectum. Finally, the disease spectrum observed in this family is reminiscent of hereditary non-polyposis colorectal cancer (HNPCC) illustrating that even though the basis of the two diseases is different, presentation can be remarkably similar. In the other two exon 9 mutation positive families the disease phenotype is consistent with that reported in families harbouring other mutations in this exon, adding further evidence to the notion that these mutations are associated with an attenuated phenotype.
Mutations identified in the APC gene
Disease characteristics of the mixed hyperplastic/adenomatous polyposis group B families
Disease characteristics of the colonic adenomatous APC mutation negative families
The second mutation of interest resulted in a stop codon at position 1969 making it one of the few 3′ mutations identified to date. The genetic change is not a simple one as it involved the duplication of a 17 bp sequence, which results in a nonsense chain of 39 amino acids prior to a termination codon. This change results in an increased propensity to develop extracolonic disease (most notably desmoid disease and osteomas), which appears to be a consistent finding with 3′APC gene mutations. Similar to other 3′APC mutations, the associated colonic phenotype is highly variable; some patients presented with typical symptoms of disease whereas others presented with extremely mild colonic features. The genotype/phenotype correlation in this family is consistent with that observed in other families harbouring 3′APC mutations.14 ,31 ,32 This family provides further evidence that 3′ APCmutations have a distinctive phenotype whereas mouse models of 3′Apc mutations suggest otherwise.33 A major difference between this set of families and other series that have been reported in the literature was the male to female ratio. Given the size of the population under investigation we believe this to be due to chance and not a real difference between this population and others that have been investigated elsewhere.
Several families were identified that, using our detection strategy, did not harbour germline mutations in theAPC gene but did present with a colonic phenotype which was consistent with a diagnosis of either FAP or attenuated FAP. Clinical features of these families were compared with those found in families where an APCgermline mutation was identified. From the families that did not harbour detectable germline APC mutations two groups could be clearly identified. Group A consisted of patients who presented with only colonic adenomatous polyposis (albeit towards the lower range, 20 to several hundreds of adenomas). This group also tended to be older at diagnosis of colorectal cancer compared with group B and the mutation positive families and it affected slightly more males than females. Group B consisted of affected individuals harbouring a mixed hyperplastic/adenomatous polyposis phenotype who were older at diagnosis compared with the APC mutation positive group but not group A. Age at diagnosis is expected to be older in groups A and B by virtue of the fact that families clearly fulfilling the diagnostic criteria for FAP were more likely to be entered into a colonic screening programme at an earlier age compared with those in groups A and B. Notwithstanding, the average age at diagnosis of FAP occurs in the fifth decade of life, whereas the average age of diagnosis was in the sixth decade of life for group B, indicative of a real difference between the two groups. Group A tended to present at an age which was similar to that of FAP reported in the literature.34
Little is known of the aetiology of hyperplastic polyps or their relationship to adenomatous polyps. Recent reports have suggested that they may represent a distinct entity that is associated with an increased risk of colorectal cancer which is greater than the population risk but less than that observed for predispositions35 such as FAP or HNPCC. The numbers of polyps in this group were lower than group A (range 20–70). Unfortunately, it was not possible to obtain the ratio of hyperplastic to adenomatous polyps. Similarly, age at diagnosis was older than the mutation positive group but was also younger than the average age in group A. This suggests that there are at least two groups of polyposis patients not linked to the APC gene.
The diagnosis of mixed adenomatous/hyperplastic polyposis in oneAPC gene positive family may be related to the family specific mutation in exon 14 of theAPC gene. Furthermore, it cannot be ruled out that the hyperplastic polyp is a somatic variant occurring on a genetic background of colorectal cancer risk. In contrast, the nine families that comprised group B where no APCmutations were identified presented with only a colonic phenotype of hyperplastic/adenomatous polyposis. Additional evidence to suggest that the two entities are distinct was the presence of extracolonic disease in the adenomatous group (desmoid disease) whereas only colonic disease was observed in the mixed hyperplastic/adenomatous polyposis group.
In summary, the mutation detection strategy used in this report will minimise the number of false negative families screened using the protein truncation test assay. Accurate description of polyposis families that do not harbour APC germline mutations will aid in classifying unique features that could help in the identification of additional genes related to at least one other genetic entity associated with colonic polyposis.
Acknowledgments
This work was supported in part by the Hunter Area Pathology Research Grant and the Hunter Area Health Service. We would like to thank all clinicians and patients who provided us with information so that this study could be performed.
Abbreviations used in this paper
- FAP
- familial adenomatous polyposis
- APC
- adenomatous polyposis coli
- AAPC
- attenuated adenomatous polyposis coli
- CHRPE
- congenital hypertrophy of the retinal pigment epithelium
- SSCP
- single strand conformation polymorphism
- DGGE
- denaturing gradient gel electrophoresis
- PTT
- protein truncation test
- ARMS
- amplification refractory mutation system
- HNPCC
- hereditary non-polyposis colorectal cancer
- PCR
- polymerase chain reaction
References
Footnotes
↵† The Hunter Family Cancer Service includes Claire Groombridge, Bronwyn Burgess, Anne Hammond, and Gillian Turner