Article Text

Abstract
BACKGROUND Leptin regulates feeding behaviour and therefore may be a mediator of anorexia associated with acute and chronic inflammation. Recently, leptin mRNA and leptin protein were found in the gastric epithelium.
AIM The aim of the present study was to examine the effect ofHelicobacter pylori infection on gastric leptin expression to investigate the pathophysiological role of gastric leptin.
METHODS Surgically resected human stomach tissues were subjected to immunohistochemistry and reverse transcriptase-polymerase chain reaction (RT-PCR) to check for the presence of leptin in the human gastric epithelium. A total of 201 H pyloripositive patients with chronic gastritis underwent eradication therapy for H pylori and were examined for the effect of infection cure in terms of body mass index (BMI) and serum leptin levels. Biopsy specimens from the gastric fundic mucosa were obtained from 40 of the 201 patients before and three months after eradication therapy. These samples were subjected to quantitative RT-PCR to examine the effect of eradication therapy on leptin expression in the gastric fundic mucosa.
RESULTS Leptin immunoreactive cells were detected in the lower half of the gastric fundic glands and a leptin PCR product was also found in the gastric fundic mucosa. H pylori infection significantly increased gastric leptin expression. In addition, cure ofH pylori infection significantly reduced gastric leptin expression, with a concomitant increase in BMI. In contrast, serum leptin levels did not change significantly after cure of H pylori infection.
CONCLUSION Leptin is present in the human gastric mucosa. Gastric leptin may play a role in weight gain after eradication of H pyloriinfection. Gastric leptin may have a local rather than systemic action.
- Helicobacter pylori
- leptin
- gastric mucosa
- body mass index
- gastritis
Abbreviations used in this paper
- RT-PCR
- reverse transcriptase-polymerase chain reaction
- CCK
- cholecystokinin
- BMI
- body mass index
- UBT
- urea breath test
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- TNF
- tumour necrosis factor
- IL
- interleukin
Statistics from Altmetric.com
Leptin, a product of the ob gene, is a 16 kDa protein synthesised by adipose tissue that plays a crucial role in the homeostasis of body weight by regulating food intake and energy expenditure. Fasting and starvation decrease leptin mRNA and protein levels, and this substantial fall produces a signal to eat.1-4 Leptin was initially reported to be secreted only by adipocytes and the placenta.1 ,5 Recently, Badoet al reported that leptin mRNA and leptin protein were found in the rat gastric epithelium, and that cells in the glands of the gastric fundic mucosa were immunoreactive for leptin.6 They also reported that both feeding and administration of CCK-8 (the biologically active carboxy terminal end of cholecystokinin (CCK)) resulted in a rapid and marked decrease in both leptin cell immunoreactivity and leptin concentration in the fundic epithelium, with a concomitant increase in the concentration of leptin in plasma, suggesting that gastric leptin may be involved in early CCK mediated effects activated by food intake, possibly including satiety. In contrast, anorexia and loss of lean body mass are hallmark manifestations of acute or chronic disease, including infection or cancer.7-11 Helicobacter pylori infection is a cause of chronic active gastritis.12 ,13 H pylorigastritis causes alarm symptoms such as anaemia, weight loss, and dysphagia in an as yet undefined subset of infected subjects.14-16 It has also been reported thatH pylori infection influences growth rate in children.17 ,18 In addition, weight gain after curingH pylori infection has been reported.19 It is possible that gastric inflammation induced by H pylori infection affects expression of gastric leptin which may play a role in the regulation of food intake. However, the relationship between H pylori infection and gastric leptin expression has not been examined.
Therefore, we examined the effect of H pylori infection on gastric leptin expression to investigate the pathophysiological role of gastric leptin.
Methods
EXPERIMENTAL DESIGN
Firstly, surgically resected human stomach tissue samples obtained from early gastric cancer patients (two cases; a 56 year old man and a 52 year old woman) were subjected to immunohistochemistry and reverse transcriptase-polymerase chain reaction (RT-PCR) to check for the presence of leptin in the gastric epithelium.
Secondly, 201 H pylori positive patients with chronic gastritis (121 men and 80 women, mean age 41.5 years) were chosen from subjects who visited multiphasic health testing services held by the Second Department of Internal Medicine, Fukui Medical University. The services included upper gastrointestinal endoscopy or radiography to screen for gastric cancer. Chronic gastritis was diagnosed using these examinations.20 ,21 Patients who had a history of peptic ulcers, reflux oesophagitis, malignant diseases, gall stones, or surgery, as well as those receiving non-steroidal anti-inflammatory drugs or antibiotics were excluded from the study.H pylori infection was diagnosed using the13C urea breath test (UBT). Subjects fasted overnight prior to the UBT. Breath samples were collected before and 15 minutes after intake of a 13C urea solution (100 mg 13C urea in 100 ml water) and analysed using isotope ratio mass spectrometry. A rise in the delta value of 13CO2 of more than 2.5/ml over baseline values was considered positive forH pylori infection. A standardised questionnaire was used to assess abdominal symptoms. This questionnaire addressed seven persistent or recurrent symptoms for more than three months which served as the primary target variables (upper abdominal pain, heart burn, regurgitation, nausea, vomiting, upper abdominal fullness, loss of appetite). The intensity of the respective symptoms was scored as 0 (absent), 1 (low intensity), 2 (moderate intensity), or 3 (severe). A total of 121 of 201 patients (60.2%) had dyspepsia symptoms. All 201 patients underwent eradication therapy forH pylori infection with omeprazole (20 mg twice daily), amoxicillin (750 mg twice daily), and clarithromycin (200 mg twice daily) for one week for gastric cancer prophylaxis. Cure ofH pylori infection was evaluated using the UBT three months after treatment. Body weights and symptom scores of the patients were measured before and three months after eradication therapy. The change in body mass index (BMI kg/m2) after eradication therapy was compared between the eradicated and non-eradicated groups. Serum samples were obtained before and three months after eradication therapy in the morning after an overnight fast, and stored at −20°C until analysed.
Thirdly, biopsy specimens from the gastric fundic mucosa were obtained before and three months after eradication therapy from 40 of 201 patients (33 patients were successfully eradicated: 20 men and 13 women, mean age 42.2 years; and seven patients were not eradicated: four men and three women, mean age 41.6 years). These samples were subjected to quantitative RT-PCR to examine the effect of eradication therapy on leptin expression in the gastric fundic mucosa. Biopsy specimens of the gastric fundic mucosa obtained from 20H pylori negative normal controls (13 men and seven women, mean age 40.8 years) were also subjected to quantitative RT-PCR to examine gastric leptin expression in controls.
These studies were performed according to the principles of the Declaration of Helsinki, and consent was obtained from each individual after a full description of the nature and protocol of the study.
IMMUNOHISTOCHEMISTRY FOR LEPTIN
The surgically resected tissue samples were fixed in Bouin's solution and embedded in paraffin. Immunohistochemical staining was performed using the avidin-biotin-peroxidase complex technique (Vectastain ABC kit; Vector, Burlingame, California, USA) described by Hsu and colleagues.22 All incubations were done at room temperature. Each section (4 μm thick) was immersed in 0.3% H2O2 in methanol for 30 minutes to destroy endogenous peroxidase activity, washed for 10 minutes with 0.05 M Tris buffered saline, pH 7.5, and then incubated for 30 minutes with normal goat serum to block non-specific Ig binding sites. The goat serum was removed and the slides were incubated overnight with a 1:100 dilution of the antihuman leptin rabbit antibody 18545S (IBR, Gumma, Japan). This was followed by a 30 minute wash in Tris buffered saline, 30 minute incubation with biotinylated goat antirabbit IgG, repeat wash in Tris buffered saline, and a 30 minute incubation with avidin DH biotinylated horseradish peroxidase. After a final wash in Tris buffered saline, the slides were incubated for two minutes in a solution containing 0.05% diaminobenzidine and 0.015% H2O2. Nuclear staining was carried out with haematoxylin. The specificity of the immunohistochemical reaction was first established by showing that non-immune rabbit serum did not reveal positive staining. In addition, the specificity of the antibody was evaluated by absorption controls. The diluted antibody was incubated with human leptin in concentrations of up to 10 nmol/ml for 24 hours at 4°C and then applied to the tissue section.
LEPTIN ASSAY
Serum leptin concentrations were determined using radioimmunoassay kits (Linco Research, St Charles, Missouri, USA) according to the manufacturer's protocol.23
QUANTITATIVE RT-PCR
Sections of surgically resected tissue samples of the gastric fundic, antral mucosa, and gastrocolic omentum were frozen and stored in liquid nitrogen. mRNA was purified from tissues using Micro FastTrack kits (Invitrogen, Carlsbad, California, USA) according to the manufacturer's protocol.24 RT-PCR for leptin was done using a novel real time quantitative RT-PCR method using an ABI 7700 sequence detector system (PE Applied Biosystems, Foster City, California, USA).25 The sense primer for leptin was 5′-CACCAAAACCCTCA TCAAGACA-3′ and the antisense primer was 5′-CTTTCTGTTTGGAGGAGACTGACT-3′. The reaction mixture was prepared according to the manufacturer's protocol using TaqMan Gold RT-PCR Kits (PE Applied Biosystems). The reactions also contained the target hybridisation leptin probe (5′-CACCA GGATCAATGACATTTCACACACG-3′). The probe was labelled with a reporter fluorescent dye 6-carboxyfluorescein at the 5′ end. The thermal cycling conditions included 48°C for 30 minutes and 95°C for 10 minutes, followed by 50 cycles of amplification at 95°C for 15 seconds and 60°C for one minute. The PCR products were also examined by 2% agarose gel electrophoresis. The gels were stained with ethidium bromide to confirm successful amplification of the expected sequences. mRNA was also purified from biopsy specimens and subjected to real time quantitative RT-PCR for measurement of leptin and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as controls using TaqMan GAPDH control reagents (PE Applied Biosystems). For relative quantification of leptin expression, a calibration curve was constructed using the mRNA obtained from the gastrocolic omentum. RNA for GAPDH was used as an endogenous control. The amount of leptin RNA was normalised to the level of GAPDH.
STATISTICAL ANALYSIS
Results are expressed as mean (SD). The significance of differences between groups for mean age, BMI, and change in BMI was evaluated using an unpaired Student's ttest. Comparison of differences in BMI changes of more than 0.5 kg/m2 was performed using a χ2 test with Fisher's exact probability test. Differences in serum leptin concentrations and relative RNA quantification of leptin in the interval before and after eradication therapy were analysed with a paired Student's t test. Symptom scores among groups were compared by Wilcoxon's signed rank test. The relationship between the change in BMI and the change in symptom scores or the change in relative RNA quantification of leptin was evaluated by Spearman's rank correlation. Differences with a p value of less than 0.05 were considered significant.
Results
DETECTION OF LEPTIN IN THE GASTRIC MUCOSA
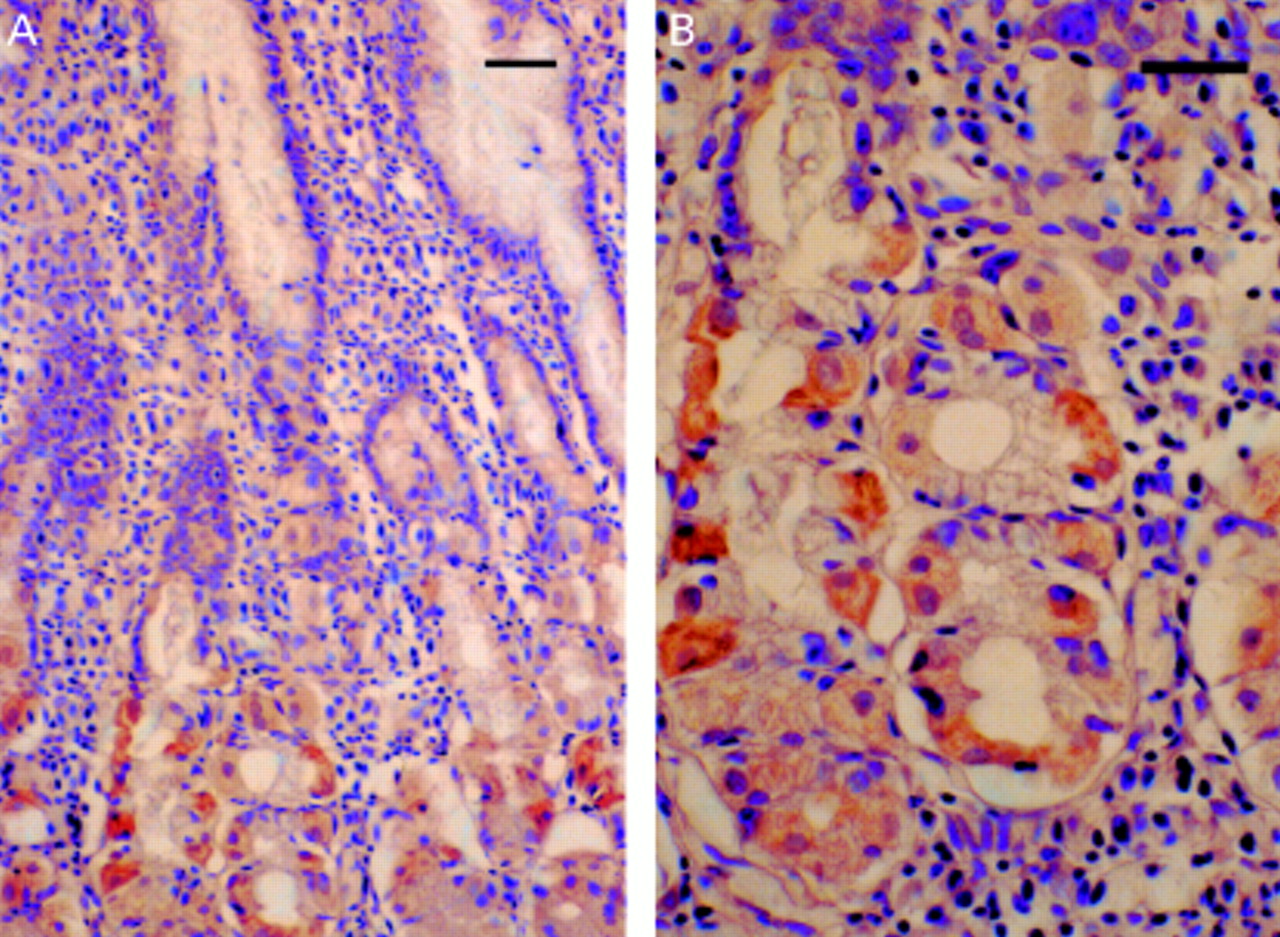
Leptin immunoreactive cells were seen in the lower half of the fundic epithelial glands (fig 1). No immunoreactivity was seen after adsorption of the antibody with 10 nmol/ml of a leptin fragment. A leptin PCR product was found in the fundic epithelium (fig 2).

Immunostaining of leptin in the human fundic mucosa. Leptin immunoreactivity was seen in the lower half of the fundic epithelial glands (A). Higher magnification (B). Scale bars 10 μm.

Reverse transcriptase-polymerase chain reaction analysis of leptin mRNA: negative control (lane 1), gastrocolic omentum (lane 2), and fundic mucosa (lane 3).
EFFECTS OF ERADICATION THERAPY ON CHANGE IN BMI
The change in BMI after eradication therapy was compared between the eradicated and non-eradicated groups. H pylori infection was cured in 161 cases. The mean change in BMI was significantly higher in the successfully eradicated patients compared with that in non-eradicated patients. A BMI change of more than 0.5 kg/m2 was observed significantly more frequently in the eradicated patients than in the non-eradicated patients (table1).
Effect of Helicobacter pylori eradication therapy on body mass index (BMI)
SERUM LEPTIN CONCENTRATION
Serum leptin levels did not change significantly after eradication therapy in either the eradicated (3.5 (2.2) to 3.9 (2.6) ng/ml) or non-eradicated (3.3 (2.6) to 3.8 (2.9) ng/ml) patients groups.
SYMPTOM SCORES
Symptom scores improved significantly after successful eradication therapy. In contrast, in the non-eradicated patients, symptom scores did not improve significantly after therapy (table 2). It is possible that improvement in symptoms after cure of H pylori infection caused the weight gain. However, there were no significant correlations between the change in symptom score and the change in BMI (fig 3).
Change in symptom scores before and after eradication therapy

Relationship between the change in symptom score and change in body mass index (BMI). There was no significant correlation (r=0.10, p=0.189).
QUANTITATIVE ANALYSIS OF GASTRIC LEPTIN
Relative RNA quantification of gastric leptin inH pylori positive patients with chronic gastritis (2.3 (0.6)) was significantly higher than that inH pylori negative normal controls (1.2 (0.5)) (p<0.0001). Relative RNA quantification of leptin significantly decreased after successful eradication. However, levels did not change significantly in the non-eradicated patients (fig 4, table 3). There was a significant correlation between the change in the relative RNA quantification of leptin and the change in BMI (r=−0.69, p=0.0002) (fig 5).

A calibration curve was constructed using four different dilutions of the mRNA obtained from the gastrocolic omentum. (A) Amplification plots of leptin; (B) calibration curve for leptin.
Effect of Helicobacter pylori eradication therapy on expression of gastric leptin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Relationship between the change in relative RNA quantification of gastric leptin and the change in body mass index (BMI). There was a significant correlation between the two (r=−0.69, p=0.0002).
Discussion
Leptin has been reported to be secreted only by adipocytes and the placenta.1 ,5 Bado et al first reported that leptin mRNA and leptin protein were present in the rat gastric epithelium, and that cells in the glands of the gastric fundic mucosa were immunoreactive for leptin.6 Recently, immunoreactive leptin cells were found in the lower half of the gastric fundic glands in humans, and leptin mRNA was also detected in human gastric mucosa by RT-PCR.26 ,27 In the present study, we confirmed the presence of leptin in the human gastric fundic mucosa. The physiological function of gastric leptin is unknown. Badoet al reported that both feeding and administration of CCK-8 resulted in a rapid and large decrease in both leptin cell immunoreactivity and the leptin content of the fundic epithelium, with a concomitant increase in the concentration of leptin in plasma. They suggested that gastric leptin may be involved in early CCK mediated effects activated by food intake, possibly including satiety.6
In contrast, anorexia and loss of lean body mass are hallmark manifestations of acute or chronic disease, including infection or cancer. The role of tumour necrosis factor (TNF), interleukin (IL)-1, and IL-6 as endogenous mediators of the host response to infection or malignancy has been extensively studied.7-9 It has also been reported that multiple cytokines and inflammation raise leptin levels. Leptin regulates feeding behaviour and therefore may be a mediator of anorexia associated with acute and chronic inflammation.28-30 Several studies have shown the involvement of cytokines in the pathogenesis of gastric inflammation. Crabtree et al detected higher levels of TNF-α, IL-6, and IL-8 in the culture supernatants ofH pylori infected gastric biopsy specimens than in specimens from uninfected patients.31 Noachet al also detected increased levels of IL-1β, IL-8, and TNF-α in culture supernatants of antral biopsy specimens from H pylori infected patients.32 Yamaoka et alreported that expression levels of IL-6, IL-7, IL-8, and IL-10 mRNA were significantly higher in mucosa with H pylori positive gastritis than in H pylori negative normal mucosa, and that this increased cytokine expression due to H pylori infection was reduced significantly after H pyloriinfection was cured.33 In the present study,H pylori infection increased gastric leptin expression, and cure of H pylori infection induced a reduction in gastric leptin expression. These findings suggest that gastric inflammation induced by H pylori infection raises gastric leptin expression.
Interestingly, cure of H pylori infection also induced significant weight gain, and there was a significant correlation between the change in relative RNA quantification of gastric leptin and change in BMI. Gastric leptin may play a role in weight gain after cure of H pyloriinfection. However, serum leptin levels did not change significantly after cure of H pylori infection. Gastric leptin may have a local rather than a systemic action, exerting paracrine effects within the gastric mucosa or stimulating vagal afferents to signal the central nervous system. Recently, several investigators reported the presence of leptin and its receptors in the human stomach, and suggested a paracrine and/or autocrine effect of leptin on gastric epithelial cell function.26 ,27 In addition, leptin sensitive vagal afferent terminals have been identified in the rat stomach.34 On the other hand, symptom scores improved significantly after successful eradication therapy. It is possible that the improvement in symptoms, rather than a change in gastric leptin expression, caused weight gain. However, there was no significant correlation between the change in BMI and the change in the dyspepsia symptom score.
In conclusion, gastric inflammation induced by H pylori infection raised gastric leptin expression. Cure ofH pylori infection reduced gastric leptin expression, with a concomitant increase in BMI. Gastric leptin may play a role in weight gain after curing H pyloriinfection. Evidence for the presence of leptin in the gastric fundic mucosa opens new avenues in the study of the regulatory mechanism of satiety. Further examination of the physiological role of gastric leptin is warranted.
Acknowledgments
This work was supported by a grant in aid for scientific research (11670555) from the Ministry of Education, Science, Sports, and Culture, Japan.
Abbreviations used in this paper
- RT-PCR
- reverse transcriptase-polymerase chain reaction
- CCK
- cholecystokinin
- BMI
- body mass index
- UBT
- urea breath test
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- TNF
- tumour necrosis factor
- IL
- interleukin