Article Text

Abstract
Background—The “topical” effect of non-steroidal anti-inflammatory drugs (NSAIDs) seems to be an important cause of NSAID induced gastrointestinal damage.
Aim—To examine the possible mechanism of the “topical” phase of damage in the small intestine.
Methods—Electron microscopy and subcellular organelle marker enzyme studies were done in rat small intestine after oral administration of indomethacin (doses varied between 5 and 30 mg/kg). The effect of conventional and non-acidic NSAIDs on rat liver mitochondrial respiration was measured in vitro in a Clarke-type oxygen electrode.
Results—The subcellular organelle marker enzymes showed mitochondrial and brush border involvement within an hour of indomethacin administration. Electron microscopy showed dose dependent mitochondrial changes following indomethacin administration consistent with uncoupling of oxidative phosphorylation (or inhibition of electron transport) which were indistinguishable from those seen with the uncoupler dinitrophenol. Parenteral indomethacin caused similar changes, but not in rats with ligated bile ducts. A range of NSAIDs, but not paracetamol or non-acidic NSAIDs which have a favourable gastrointestinal tolerability profile, uncoupled oxidative phosphorylation in vitro at micromolar concentrations and inhibited respiration at higher concentrations. In vivo studies with nabumetone and aspirin further suggested that uncoupling or inhibition of electron transport underlies the “topical” phase of NSAID induced damage.
Conclusion—Collectively, these studies suggest that NSAID induced changes in mitochondrial energy production may be an important component of the “topical” phase of damage induction.
- intestinal inflammation
- intestinal toxicity
- mitochondrial function
- drug induced toxicity
Statistics from Altmetric.com
Non-steroidal anti-inflammatory drugs (NSAIDs) are the most prescribed drugs worldwide which attests to their efficacy as analgesics, and anti-inflammatory and anti-pyretic agents. The main concern with this group of drugs is the frequency and severity of their gastrointestinal side effects.1 ,2
The pathogenesis of intestinal damage is incompletely understood. It is generally accepted that the early pathogenic events include a “topical” phase in addition to the inhibition of cyclooxygenase, followed by a multistage pathogenic event in which intestinal permeability, luminal contents, neutrophils, and the microcirculation all play a role in the development of inflammation and ulcers.3-6 The importance of inhibition of cyclooxygenase in the damage induced by NSAIDs has been clearly documented.3 Nevertheless, it is it is possible to inhibit cyclooxygenase selectively without inducing intestinal damage.7-9 Furthermore, disruption of the prostaglandin synthase-1 gene coding for cyclooxygenase-1 in mice, with resulting mucosal prostaglandin concentrations of less than 1% of that of controls, is not associated with gastrointestinal pathology.10 In the stomach changes in drug formulation (enteric coating, rectal administration, etc.)9 ,11 and abolishing gastric acid secretion (abrogates aspirin (pKa 3.5) induced damage12 presumably by limiting the gastric absorption of the drug) greatly increase tolerability of NSAIDs in the short term. This suggests that the “topical” action of NSAIDs may be an important co-factor in the initiation of the damage.3 The “ion trapping” hypothesis which postulates that accumulation of NSAIDs in intestinal epithelial cells depends largely on the interaction of the acidity of the NSAID (pKa) and luminal pH provides a basis for (along with molecular size, lipid solubility, contact time, etc.) the “topical” action of NSAIDs but not the mechanism.13 ,14
One suggestion is that uncoupling of oxidative phosphorylation may be the biochemical mechanism underlying the “topical” toxicity of NSAIDs.5 ,6 ,15 Uncoupling of oxidative phosphorylation has been documented in vitro with some NSAIDs3 ,16 ,17 and in vivo in the case of aspirin.18
The aim of this study was to examine the possible mechanism of the “topical” phase of NSAID induced gastrointestinal damage in the rat by using electron microscopy and subcellular organelle marker enzyme studies.
Material and Methods
ANIMALS
Male pathogen-free Sprague-Dawley rats (Charles River), 6–8 weeks old, weighing 200–250 g, were used throughout these studies. For documentation and confirmation of the ulcer location and frequency, histology, subcellular organelle marker enzyme studies and electron microscopy, animals were fasted overnight before receiving NSAIDs or solvent by gastric gavage via a soft rubber tube (without sedation) at 0800 or 1200 hours. The 0800 hours group of animals remained fasted and were anaesthetised (with Hypnovel-Hypnorm) 1–6 hours later, underwent laparotomy and were killed by cervical dislocation after the procedure. In the case of the animals receiving the drugs at noon, these were re-fed (two hours later) and operated on the next day 20 hours later (with a preceding overnight fast).
For the in vivo studies indomethacin (5, 10, 20 or 30 mg/kg) was dissolved in a bicarbonate buffer (pH 7.4) prior to gastric gavage. The range of indomethacin doses used was similar to that used in most other comparable studies when assessing the acute gastrointestinal toxicity of NSAIDs. These doses are somewhat higher than that required to demonstrate anti-inflammatory activity in carrageenen paw oedema (2.5 mg/kg). Aspirin (100 mg/kg) and nabumetone (600 mg/kg) were initially dissolved in 10% dimethylsulphoxide (DMSO) and diluted to 1 ml just prior to administration (final concentration of DMSO <5%) and was followed by 1 ml water. The doses of aspirin and nabumetone are roughly equivalent to about eight and 21 times the maximum amount given to a 70 kg person with rheumatoid arthritis, respectively (assuming 3–4 g/day for aspirin and 2 g/day nabumetone). The equivalent dose of indomethacin, calculated in the same way, is 2.5-fold (5 mg/kg dose) to 14-fold (30 mg/kg dose) higher. Dinitrophenol (0.4 ml of a 6 mM solution) was dissolved in 10% DMSO and administered at a final concentration of less than 2% DMSO. The same amount of solvent was used in control experiments.
SUBCELLULAR ORGANELLE MARKER ENZYME STUDIES
Groups of eight to 24 rats received indomethacin (30 mg/kg) or solvent by gavage and underwent laparotomy one, six, or 20 hours later. Samples from the duodenum (third part), jejunum (30 cm distal from the ligament of Treitz) and ileum (10 cm proximal to the caecum) were minced finely in a Polytron homogeniser (Kinematica GmbH, Lucern, Switzerland) for 15 seconds at speed setting 1 and disrupted by 15 strokes of a loose fitting (type A) pestle in a Dounce homogeniser in (10% w/v) ice cold 0.25 M sucrose containing disodium EDTA (1 mmol/l, pH 7.4) and ethanol (22 mmol/l). Homogenates were stored at −70°C until assay of enzyme activities.
Organelle specific marker enzymes studies were carried out as described previously19 ,20: alkaline phosphatase (EC 3.1.31), brush border; lactate dehydrogenase (EC 1.1.1.27), cytosol; DNA, nucleus; succinate dehydrogenase (EC 1.3.99.1), inner mitochondrial membrane; citrate synthase (EC 4.1.3.7), mitochondrial matrix (tricarboxylic acid cycle); cytochrome c oxidase (1.9.3.1), mitochondrial matrix (electron transport chain); b-N-acetyl-D-glucosaminidase (EC 3.2.1.30), lysosomes; α-glucosidase (EC 3.2.1.20), endoplasmic reticulum.
Mitochondrial enzymes were assayed after freeze thawing three times to disrupt the mitochondrial membranes.
Protein content was analysed according to Lowry et al,21 and DNA content according to Carter et al.22
MORPHOLOGY AND ELECTRON MICROSCOPY
A straightforward small intestinal ulcer count was made, following exposure of the mucosa by a longitudinal cut along the mesenteric border, 20 hours after administration of the drugs.
For microscopy an abdominal incision was made under anaesthesia, the stomach was opened and a catheter placed in the first part of the duodenum. The whole of the small intestine was then flushed, avoiding distension, with a 3.0% gluteraldehyde in 0.1 M sodium phosphate buffer pH 7.3–7.4. One centimetre lengths of duodenum (third part), jejunum (30 cm distal to the ligament of Trietz) and ileum (10 cm proximal to the caecum) were then processed: (i) to paraffin wax blocks and 3–4 μm sections were cut and stained with haematoxylin and eosin (six animals each studied as controls and at one, six, or 20 hours after administration of indomethacin 20 mg/kg); or (ii) for electron microscopy and placed in gluteraldehyde for three days (four animals as controls and at one, two, or five hours after indomethacin 20 mg/kg was given). A further four animals in each group were studied by electron microscopy (jejunum only) one hour after instillation in the small intestine of 0.4 ml of a 6 mM solution of dinitrophenol, a potent uncoupler of oxidative phosphorylation, and two hours after administration of 5 and 10 mg/kg indomethacin by gastric gavage to assess a possible dose response relation.
After fixation, samples for electron microscopy were embedded in araldite and ultrathin sections were cut using an Ultratome-Richart Ultracut-E. The samples were then examined with a Joel 1200 cm electron microscope in transmission mode. All experiments were carried out without knowledge of treatment.
IN VITRO MITOCHONDRIAL STUDIES
Animals were fasted overnight. Rat liver mitochondria were isolated and used for these studies as it is exceedingly difficult to obtain high yield, purified coupled mitochondria from intestinal tissue.23 However, it is generally accepted that there are no major differences between mitochondria isolated from different organs with regard to responses to uncoupling agents or inhibitors of the respiratory chain.23 After cervical dislocation livers were rapidly removed and placed in ice cold 0.9% saline. The liver was then cut into approximately 1 cm3 pieces and homogenised in 50 ml of a 74 mM sucrose, 225 mannitol, 1 mM EDTA, 5 mg/ml bovine serum albumin solution in 10 mM MOPS-NaOH buffer, pH 7.4, by six strokes of a tight fitting Teflon pestle. The homogenate was centrifuged at 500 g for 10 minutes and the resulting supernatant re-centrifuged at 12 000 g for 10 minutes. The pellet was resuspended in the sucrose solution and centrifuged for 10 minutes at 12 000 g to give the resulting mitochondrial enriched pellet used for these experiments. All procedures were performed at 4°C.
Oxygen consumption was measured in a Clarke-type oxygen electrode, as described previously.24 The reaction mixture consists of 150 mM sucrose, 10 mM potassium chloride, 5 mM magnesium chloride, and 1 mM potassium dihydrogen orthophosphate in 10 mM HEPES-NaOH buffer, pH 7.4. Substrates, inhibitors and drugs were introduced into the chamber (1.0 ml capacity) by syringe and reactions were carried out at 30°C under magnetic stirring.
Electron transport experiments were done with mitochondria after addition of 0.1 mM cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) prior to the addition of the drugs.
Rotenone and FCCP were dissolved in ethanol whereas the rest were solubilised in 10% DMSO prior to dilution and pH adjusted to 7.4. The solvent was used by itself in the control experiments. The chemical structures of the drugs used in these experiments are shown in fig 1. The conventional NSAIDs studied were indomethacin (pKa 4.5), naproxen (pKa 4.2), aspirin (pKa 3.5), flurbiprofen (pKa 4.2), and piroxicam (pKa 6.3); paracetamol was studied as a non-NSAID analgesic control. Three non-acidic NSAIDs were also studied: nabumetone (and its active acidic metabolite 6-methoxy-2-naphthylacetic acid (6-MNA; pKa 4.5)) and two chemical modifications of flurbiprofen, namely NO-flurbiprofen (fig1), a nitroxybutylester derivative of flurbiprofen, and a flurbiprofen dimer where a molecule of flurbiprofen is linked to another flurbiprofen molecule via an acid anhydrite bond. Each drug was tested over a range of concentrations and the data presented represent the mean of three to five experiments performed on different days.

: Chemical structure of the NSAIDs and paracetamol used in the study.
Mitochondrial protein was measured by using Pierce’s BCA protein assay kit, using bovine serum albumin as standard protein.
BILE DUCT LIGATION EXPERIMENTS
Pharmacokinetics permits three possible routes by which NSAIDs may come into contact with the small intestinal mucosa. Firstly, the “topical” phase following ingestion (during absorption), secondly, a systemic route as the drug enters the vascular compartment and is distributed throughout the body, and, thirdly, following excretion in bile which may re-expose the small intestine to the “topical” phase. In an attempt to discriminate between the “topical” effects of indomethacin on intestinal mitochondrial morphology and the systemically mediated effect, one group of rats underwent bile duct ligation while the other underwent sham operation (all animals were re-fed after the procedure). Twenty four hours later, after an overnight fast, the animals received indomethacin 20 mg/kg intraperitoneally. The animals whose bile ducts had been ligated also received chenodeoxycholic acid by gavage (1 ml of a 10 μmolar solution) the chief constituent of bile acid in the rat. This is an important control as bile plays a pathogenic role in indomethacin induced damage in the rat.25 ,26 Electron microscopy (jejunum only) was done two hours later on groups of four animals.
ASPIRIN EXPERIMENTS
Of the NSAIDs studied in the rat, aspirin by gavage (and intraperitoneally)8 is the only conventional NSAID, apart from nabumetone,27 that does not cause small intestinal ulcers. Aspirin and nabumetone (6-MNA) are also not excreted in significant amounts in bile following parenteral administration.28 The lack of small intestinal toxicity of aspirin is puzzling.3 One possibility is that the small intestine may not exposed to significant “topical” concentrations because of rapid absorption from the gastroduodenal mucosa (and the lack of excretion in bile).14
To study this possibility, rats underwent laporatomy under anaesthesia. One group received 100 mg/kg (volume of 1 ml) aspirin directly into the small intestine whereas the other group received the same dose by gastric gavage. Electron microscopy (jejunum only) was done two hours later on groups of four animals and an ulcer count at 20 hours (six animals in each group).
Results
INDOMETHACIN AND SMALL INTESTINAL LESIONS
Macroscopic, predominantly mid-small intestinal, damage occurred consistently 20 hours after gastric gavage of 20 or 30 mg/kg indomethacin in male Sprague-Dawley rats, as shown previously.29 ,30 There were 15–120 ulcers (median 49) in 26 of 30 treated animals. Light microscopy showed no histological inflammatory changes at one hour. At six hours there was a minor polymorphic infiltrate which became intense around the focal erosions and ulcers at 20 hours as described previously in much greater detail by Anthony et al.30
Indomethacin (5 mg/kg) caused small intestinal ulcers in two of eight animals (numbers 3 and 12) whereas seven (median 22, range 2–56) of 10 animals receiving the 10 mg/kg dose had ulcers.
SUBCELLULAR ORGANELLE ENZYME MARKER STUDIES
After indomethacin (30 mg/kg) ingestion the most noticeable changes in subcellular organelle marker enzyme activity were found in the jejunum (table 1). There was a significant decrease in the activities of the brush border marker (alkaline phosphatase) at one, six, and 20 hours. All three mitochondrial marker enzymes were increased at one hour, persisting throughout in the case of succinate dehydrogenase and citrate synthase. Markers for other subcellular structures did not alter significantly until ulcers were evident, which presumably represents the combination of the biochemical action of indomethacin and the presence and consequence of the damage associated with the acute inflammatory cell infiltrate.
Organelle marker enzyme activities in rat jejunum
ELECTRON MICROSCOPY
Electron microscopy was carried out in groups of four rats at one, two, or five hours after gavage of 20 mg/kg indomethacin (or vehicle) on mucosa from the duodenum, jejunum and ileum. Alterations were found at each site in all the indomethacin treated animals, but were most pronounced in the duodenum and jejunum. Figure 2 shows representative and the most severe changes in mitochondrial morphology in the indomethacin (20 mg/kg) treated rats. These changes were indistinguishable from those found one hour after the administration of the uncoupler dinitrophenol, which was associated with the range of mitochondrial morphological changes as described previously in other tissues.23 Two hours after administration of 5 mg/kg indomethacin there was subtle and patchy swelling and elongation of mitochondria in two rats which increased in severity with the 10 mg/kg indomethacin dose where loss of cristae also became evident, but with only occasional vacuolisation. These findings are consistent with milder degree of uncoupling or inhibition of electron transport23 and are within the range of pathology seen with dinitrophenol.

: Representative electron transmission micrographs of rat small intestine (jejunum, 30 cm distal to the ligament of Treitz) demonstrating (A) normal enterocyte mitochondria, (B) vacuolisation of mitochondria one hour after indomethacin administration, (C) vacuolisation of mitochondria one hour after the administration of the uncoupler dinitrophenol, and (D) normal enterocyte mitochondria one hour after nabumetone administration. Note normal brush border appearances. Original magnification, ×10 000.
One hour after indomethacin (20 mg/kg) administration, there were also minor degenerative changes (dilatation and some minor vacuolisation) in endoplasmic reticulum, but the brush border and other subcellular organelles appeared normal. There was an impression that the morphological changes became more severe with time.
IN VITRO MITOCHONDRIAL STUDIES
Figure 3 shows that indomethacin, aspirin, naproxen, and piroxicam uncoupled oxidative phosphorylation of isolated rat liver mitochondria in micromolar concentrations whereas paracetamol did not. However, both paracetamol and NSAIDs at higher concentrations inhibited respiration in coupled mitochondria.

: Indomethacin, aspirin, naproxen, and piroxicam stimulate mitochondrial respiration (uncouple oxidative phosphorylation) at concentrations between 30 and 1500 μM, depending on the particular NSAID, and inhibit respiration at higher concentrations. Paracetamol, a non-NSAID analgesic control, reduces respiration, without an uncoupling effect. Each data point represents the mean (SE) of 3–5 experiments, performed on different days.
The mechanism of this inhibition was studied further. Figure 4 shows that indomethacin, naproxen, aspirin, and paracetamol inhibit both glutamate/malate and succinate (with rotenone) stimulated respiration in uncoupled mitochondria. Using sonicated mitochondria with potassium ferricyanide as an artificial electron acceptor in the presence of appropriate inhibitors (rotenone, antimycin A or cyanide)23 to define the effects on individual respiratory complexes, all the drugs inhibited electron transfer in complex I, and complexes II plus III, in a concentration dependent manner (data not shown).
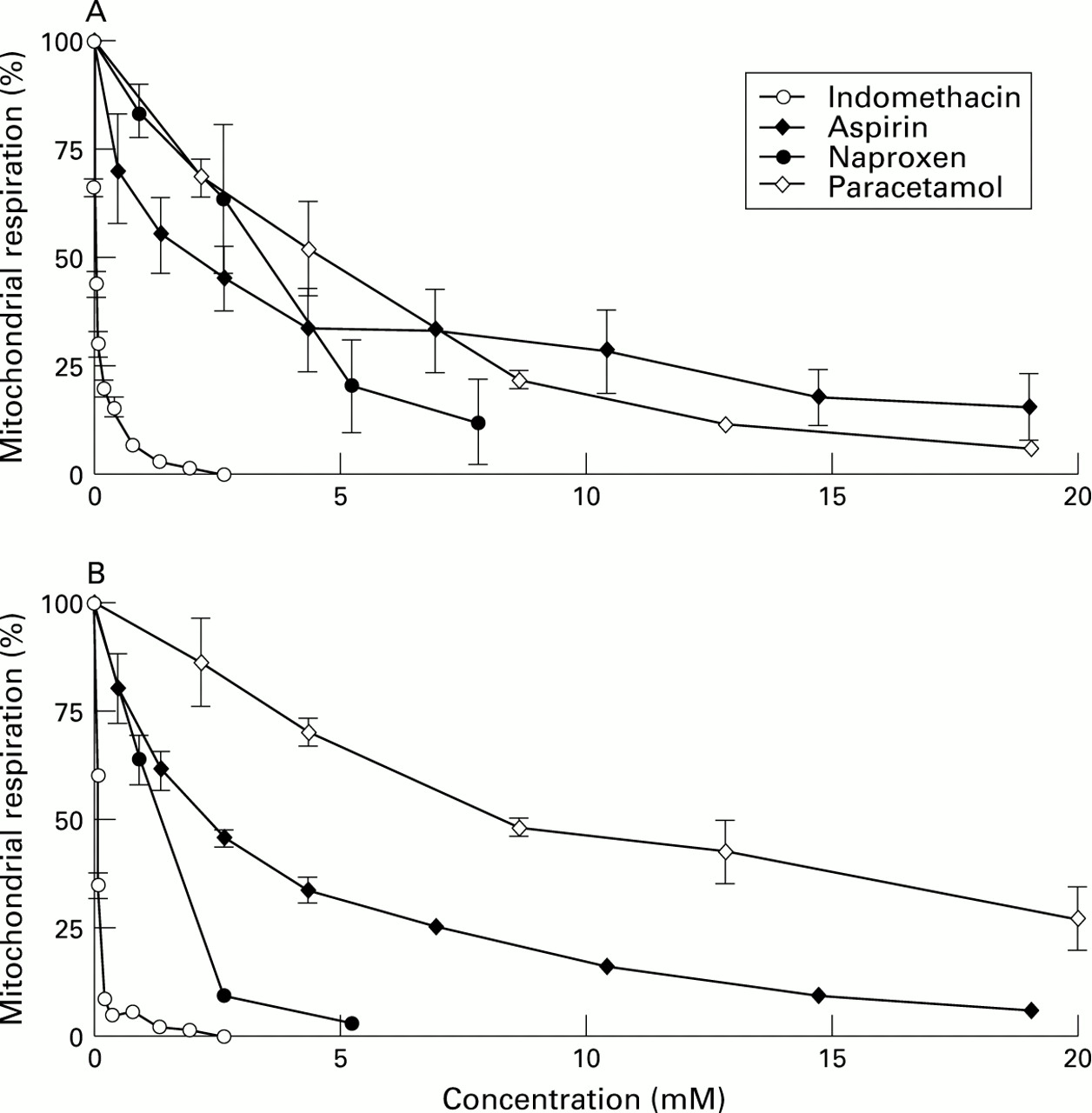
: Mitochondrial preparation was as in fig 3, but with the addition of 0.1 μM FCCP, a potent uncoupler, prior to the addition of the drugs. (A) Glutamate/malate and (B) succinate (in the presence of rotenone) were used as electron donors for complexes I and II, respectively, at different concentrations of NSAIDs as shown in the figure. Indomethacin, aspirin, naproxen, and paracetamol all inhibit complexes I and II at different concentrations in a dose dependent manner. Each point represents the mean (SE) of 3–5 separate experiments, performed on different days.
STUDIES WITH NON-ACIDIC NSAIDs
The effects of nabumetone, a non-acidic pro-NSAID, on in vitro and in vivo uncoupling and its active acidic metabolite 6-MNA in vitro, were studied.
A single 600 mg/kg dose of nabumetone by gastric gavage in eight rats did not cause macroscopic or microscopic small intestinal damage at 20 hours. Furthermore, electron microscopy of the mid-small intestine (four rats) following this dose at one and five hours showed no alterations in mitochondrial morphology (fig 2). Nabumetone also had no effect on mitochondrial respiration in isolated coupled rat liver mitochondria up to a maximum solubility (about 1.0 mM) in 10% DMSO (fig 5). The active metabolite of 6-MNA, however, increased respiration by 180 to 200% in the concentration range of 0.3–0.7 mM.

: (A) The non-acidic pro-NSAID, nabumetone, had no significant effects on mitochondrial respiration whereas its active component, 6-MNA, stimulated respiration in a similar fashion to the other NSAIDs. (B) Similarly, flurbiprofen stimulated mitochondrial respiration whereas its chemical modification to a nitroxybutyl ester or a flurbiprofen dimer, renders the drugs incapable of uncoupling oxidative phosphorylation.
Figure 5 shows that the chemically modified versions of flurbiprofen (nitroxybutyl-flurbiprofen and flurbiprofen dimer), which render them non-acidic and in the case of the former abolish gastric toxicity and reduce small intestinal damage significantly,32 had no significant effects on mitochondrial respiration.
BILE DUCT LIGATION EXPERIMENTS
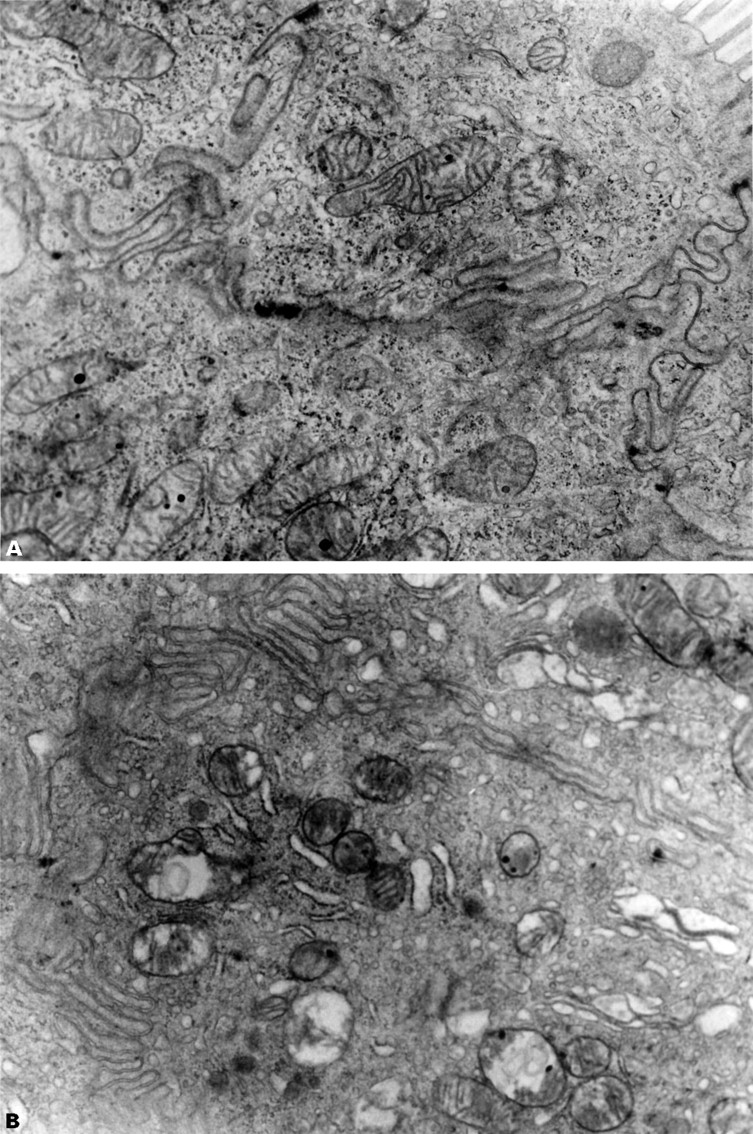
Sham operated animals with intact bile ducts, receiving indomethacin intraperitoneally, had abnormal jejunal mitochondria (representative picture shown in fig 6) resembling that seen after gavage (as above), whereas rats with ligated bile ducts had normal mitochondrial morphology. Collectively, this shows that the systemically mediated effect of indomethacin, at the doses given, is insufficient to affect small intestinal mitochondrial morphology whereas the biliary excreted component seems sufficient to cause the “topical” effect.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
: Representative electron transmission micrographs of rat small intestine following intraperitoneal indomethacin (20 mg/kg) in (A) a rat with a ligated bile duct showing normal intestinal mitochondria with discrete cristae and (B) a sham operated animal showing disruption of mitochondrial cristae, vacuolisation and occasional disruption of both the outer and inner mitochondrial membranes. Endoplasmic reticulum is also dilated. Original magnification, × 30 000.
ASPIRIN EXPERIMENTS
Aspirin uncoupled in vitro (fig 3). Animals receiving aspirin by gastric gavage had normal jejunal mitochondrial morphology on electron microscopy whereas those receiving aspirin directly into the small intestine had changes identical to those following indomethacin and dinitrophenol suggestive of uncoupling of oxidative phosphorylation and/or inhibition of electron transport. None of the animals receiving aspirin by gavage had small intestinal ulcers whereas those receiving it directly into the small bowel had severe, extensive and confluent small intestinal ulceration distal to the administration site of aspirin, suggesting that the lack of apparent small intestinal toxicity of orally administered aspirin may be due to lack of the “topical” effect because of rapid absorption.14
Discussion
Here, we show directly by a subcellular marker enzyme technique that intestinal mitochondria and brush border are affected within one hour of indomethacin administration. Electron microscopy shows dose dependent mitochondrial changes that are indicative of uncoupling of oxidative phosphorylation or inhibition of electron transport. We expand on the previous in vitro studies of NSAIDs on mitochondrial energy production, show that uncoupling relates to the acidic nature of NSAIDs and that rendering NSAIDs non-acidic and ineffective as proton translocators coincides with their improved gastrointestinal tolerability. Along with the bile duct ligation and aspirin studies, this suggests that the mechanism of the “topical” phase of NSAID damage may involve uncoupling of oxidative phosphorylation and/or inhibition of electron transport.
Although the pathogenic events of NSAID induced gastrointestinal damage are controversial, there is a consensus that there is an important “topical” component of the damage both in the stomach and small intestine9 ,33-35 which is independent of NSAID action to inhibit cyclooxygenase.3 ,36 The precise nature of the “topical” phase of damage is uncertain. Firstly, one suggestion is that NSAIDs have a detergent-like action which disrupts mucus gel37 and/or cell membrane integrity.38 ,39The decreased activity of the brush border enzyme alkaline phosphatase is consistent with this suggestion, although no major structural changes were seen on electron microscopy. Secondly, acidic NSAIDs may concentrate in the mucosa (“ion trapping”),40 ,41 but how this causes damage is uncertain. The subcellular organelle marker enzyme studies suggest that mitochondria are affected following oral indomethacin. Mitochondrial enzyme activities were all significantly increased, which is, however, an unusual response to damage and not readily explained. One possibility is that substrate access to the membrane bound enzymes is facilitated via increased mitochondrial membrane permeability caused by the effect of indomethacin on mitochondrial energy production. Whatever the explanation, the electron microscopy studies showed dose dependent in vivo mitochondrial changes following indomethacin administration which are suggestive of uncoupling and/or inhibition of electron transport. The in vitro studies show that all the acidic NSAIDs uncoupled oxidative phosphorylation at concentrations which are likely to be achieved within intestinal epithelial cells following ingestion of NSAIDs, whereas paracetamol, an analgesic without gastrointestinal toxicity, and the non-acidic NSAIDs which have an improved gastrointestinal tolerability profile, did not. These findings expand on earlier studies which demonstrated that some NSAIDs uncoupled oxidative phosphorylation,15 ,42 and indirect evidence that suggested that uncoupling of oxidative phosphorylation or inhibition of electron transport may play a role in their intestinal toxicity.5 ,43 ,44 Furthermore, our studies show that the uncoupling property of NSAIDs reside within their carboxylic or enolic acid groups (along with their lipid solubility characteristics).
The doses of indomethacin used to demonstrate in vivo uncoupling in rat small intestine were 2.5- to 14-fold higher than maximum doses used in a 70 kg human. Administration of these high doses, notwithstanding differences in species susceptibility, is a compromise between the desire to use comparable pharmacological doses as in humans and those required for consistent ulcer formation in the rat. The doses of indomethacin used in this study, although an order of magnitude greater than that required to inhibit cyclo- oxygenase,45 ,46are, however, within the range conventionally used to investigate the pathogenesis of NSAID induced gastrointestinal toxicity.
The “topical” effect of NSAIDs is most pronounced at the site of drug absorption following ingestion. Absorbed NSAIDs are largely (over 99%) bound to albumin28 so that an effective concentration for mitochondrial uncoupling may not be reached in other tissues. Certainly, there was no evidence of uncoupling of small intestinal mitochondria when indomethacin was administered parenterally to rats with ligated bile ducts. However, the main objection to the importance of the “topical” phase of damage of NSAIDs comes from the observations that: (i) parenterally administered NSAIDs cause small intestinal damage47 ,48; (ii) oral aspirin does not cause small intestinal damage (which also suggests that inhibition of cyclooxygenase is not the only explanation for the damage). In respect of (i) our studies show that parenteral indomethacin uncouples intestinal mitochondrial oxidative phosphorylation or inhibits electron transport via excretion in bile. Indeed most NSAIDs, apart from the active metabolite of nabumetone, 6-MNA, and aspirin have a significant biliary excretion component27 ,49 which exposes both the stomach (reflux of duodenal contents) and the small intestine to concentrations of the drug which may be sufficiently high to uncouple, depending on the doses given. We could not demonstrate small intestinal toxicity following very large doses of nabumetone or an effect to uncouple oxidative phosphorylation in vivo or in vitro which accords with its purported favourable gastrointestinal side effect profile in humans.2 ,50-52 However, the active metabolite of nabumetone, 6-MNA, uncoupled mitochondria in vitro, which in keeping with the results of the other acidic NSAIDs. Although oral 6-MNA clearly causes gastric damage (small bowel damage was not looked for), large intravenous doses of 6-MNA cause no intestinal damage, possibly because the intestine is not exposed to intact 6-MNA27when administered by this route. Lastly, the nitroxybutyl modification of flurbiprofen or the flurbiprofen dimer renders the drug non-acidic and ineffective as a mechanical proton translocator. Wallace et al 32 showed that the former modification diminishes or abolishes the gastric toxicity of the drug. It was suggested that the basis for reduced toxicity was the release of nitrogen oxide from the molecule, but the present studies suggest that it might additionally be due to the modification of the carboxyl group, rendering the molecule inactive as an uncoupler of oxidative phosphorylation. In this context it is interesting that esterification of aspirin is associated with strikingly improved short term gastric tolerability in the rat.53 Collectively, these findings support an important pathogenic role of the “topical” action of NSAIDs in intestinal damage.
Aspirin is clearly toxic to the gastric mucosa, but it has been particularly difficult to explain why it does not cause small intestinal damage regardless of the route of administration (orally or parenterally). Here, we show that when aspirin is instilled directly into the small intestine it is associated with uncoupling of intestinal mitochondrial oxidative phosphorylation and/or inhibition of electron transport and leads to severe small intestinal ulceration, but not when given by gastric gavage. This suggests that aspirin is so rapidly absorbed by the gastroduodenal mucosa following ingestion14 and that insufficient concentrations are achieved in the more distal small intestine (along with its lack of excretion in bile) to affect mitochondria and hence it does not exert a “topical” action at this site or cause damage. Indeed the gastric “barrier breaking” effect of aspirin is only seen following ingestion and not after intravenous administration9 ,47 of moderate doses. Parenteral aspirin in large doses does, however, cause stomach damage, possibly because blood borne, highly acidic NSAIDs, accumulate in gastric mucosal cells.54 Hence, two of the main theoretical objections to the importance of the “topical” action of NSAIDs in the pathogenesis of gastrointestinal damage have been answered in these studies.
The dual action of NSAIDs to uncouple oxidative phosphorylation–inhibit electron transport (“topical” action) and inhibit cyclooxygenase (“topical” and systemic action) provides a logical explanation for the mechanism and high prevalence of gastrointestinal toxicity of these drugs. Hence, uncoupling/inhibition following indomethacin (the precise site of absorption is in part determined by size, lipid solubility and charge of the NSAID, drug formulation, gastric pH, etc.) would result in diminished cellular ATP production, cellular calcium toxicity,55 production of reactive oxygen species6 ,55 resulting in increased mucosal permeability.56-58 Increased intestinal permeability allows luminal aggressive factors access to the mucosa which results in an inflammatory reaction. The concomitant inhibition of cyclooxygenase (which occurs at picomolar concentrations of NSAIDs and hence evident irrespective of the mode of administration of the drugs), with decreased prostaglandins, may then alter local blood flow and therefore be an important cofactor in driving the inflammation to ulcers.3-6
The importance of elucidating the mechanism of the “topical” damaging effect of NSAIDs lies in the fact that it may be possible to make them safer by chemical modification. Although NSAID damage to the gastrointestinal tract is clearly a multistage pathogenic process,6 the results of this study suggests that uncoupling of oxidative phosphorylation or inhibition of electron transport by NSAIDs may be an early pathogenic event which underlies the “topical” phase of the biochemical damage. This suggests an alternative strategy for the development of “safer” NSAIDs, namely modifications of the carboxylic group common to most NSAIDs, to render the drugs ineffective as proton translocators.
Acknowledgments
6-Methoxy-2-naphthylacetic acid (6-MNA) was graciously provided by SmithKline Beecham (UK) and the nitroxybutylester derivative of flurbiprofen and the flurbiprofen dimer by Nicox, Dublin, Ireland.
SS was supported by an unconditional grant from SmithKline Beecham, USA, GS by the Helga Jonsdottir & Sigurlidi Kristjansson Memorial Fund, Iceland, AM by an MRC Clinician Scientist Fellowship, and TM by the South East London Health Authority.