Article Text

Abstract
Background—Familial adenomatous polyposis usually results in colonic polyposis with hundreds to thousands of polyps, congenital hypertrophy of the retinal pigment epithelium (CHRPE), and variable extracolonic features. Recent reports indicate that patients with distal mutations between codons 1445 and 1578 do not express CHRPE and have a high incidence of desmoid tumours.
Patients—The family studied has an unusual phenotype of sparse colonic polyposis but profuse upper gastrointestinal polyposis. Affected subjects do not have CHRPE.
Methods—The protein truncation test followed by sequencing identified a 2 base pair deletion at codon 1520 in the APC gene. This results in a frameshift creating a stop codon 13 codons downstream.
Results—This family demonstrates that sparse colonic polyposis but severe upper tract polyposis may be associated with mutations between codons 1445 and 1578.
Conclusions—Study of duodenal and colonic polyps in further cases with mutations in this region is warranted. Such mutations may preferentially cause duodenal adenomas and desmoid tumours as somatic mutations in these tumours also occur in this region, unlike colorectal tumours where somatic mutations occur more proximally. This study emphasises the importance of screening the upper gastrointestinal tract even when the colonic disease is mild.
- familial adenomatous polyposis
- duodenal polyps
- APC mutations
- colorectal polyps
Statistics from Altmetric.com
Familial adenomatous polyposis (FAP) is an inherited disorder in which hundreds to thousands of adenomatous polyps develop in the colon. Cancer is virtually inevitable unless colectomy is performed. Most individuals later develop upper gastrointestinal polyps but these often remain small and the risk of malignancy is less than in the colon.1 ,2 Many affected individuals have retinal lesions known as congenital hypertrophy of the retinal pigment epithelium (CHRPE).3
FAP is due to mutation in the APC gene. Over 300 different germline mutations have been described scattered throughout the 5′ (proximal) half of the gene.4 Mutations near codon 1309 are associated with profuse polyposis (>2000 polyps).5-7Mutations in the extreme proximal region of the gene are associated with attenuated disease with fewer than 100 polyps.8
Relatively few families have mutations 3′ (distal) to codon 1444. Recent studies report that these individuals do not express CHRPE and have a high incidence of desmoid tumours.9 ,10 The severity of polyposis in the upper gastrointestinal tract and colon was not reported. Other studies report mutations 3′ of codon 1403 resulting in unpredictable and often severe phenotypes with extracolonic features.11 ,12 In the present study, we report a family with an APC mutation in this region and an unusual phenotype.
Methods
PATIENTS
The family was referred to the Queensland FAP Register and included in the genetic testing programme. All members of the family gave written informed consent. Affected members underwent colonoscopy and upper endoscopy as clinically indicated.
LABORATORY DATA
DNA was extracted from blood by a modified salt precipitation technique.13 The APC gene was screened for mutations using the protein truncation test essentially according to van der Luitet al 14 and heteroduplex/single stranded conformation polymorphism assays according to the manufacturer’s recommendations (MDE Gel Solution, AT Biochem, USA). A polymerase chain reaction fragment of exons 15–7 was cloned into T-vectors15 and sequenced on an ABI Automated Sequencer.
Results
Figure 1 shows the pedigree. The proband (I:1) was 29 years old when she presented with rectal bleeding. Colonoscopy showed 33 polyps which were removed and histologically confirmed to be adenomas. There was no known family history of polyps or colorectal cancer. Over the next seven years, 157 polyps were removed. At age 37 she was commenced on sulindac and over the past 10 years has had only 20 colorectal polyps removed in total during annual colonoscopic examinations. Indirect ophthalmoscopy for CHRPE was negative. A histologically confirmed fibroma was removed from the axilla at age 44 but examination reveals no other features of Gardner’s syndrome.
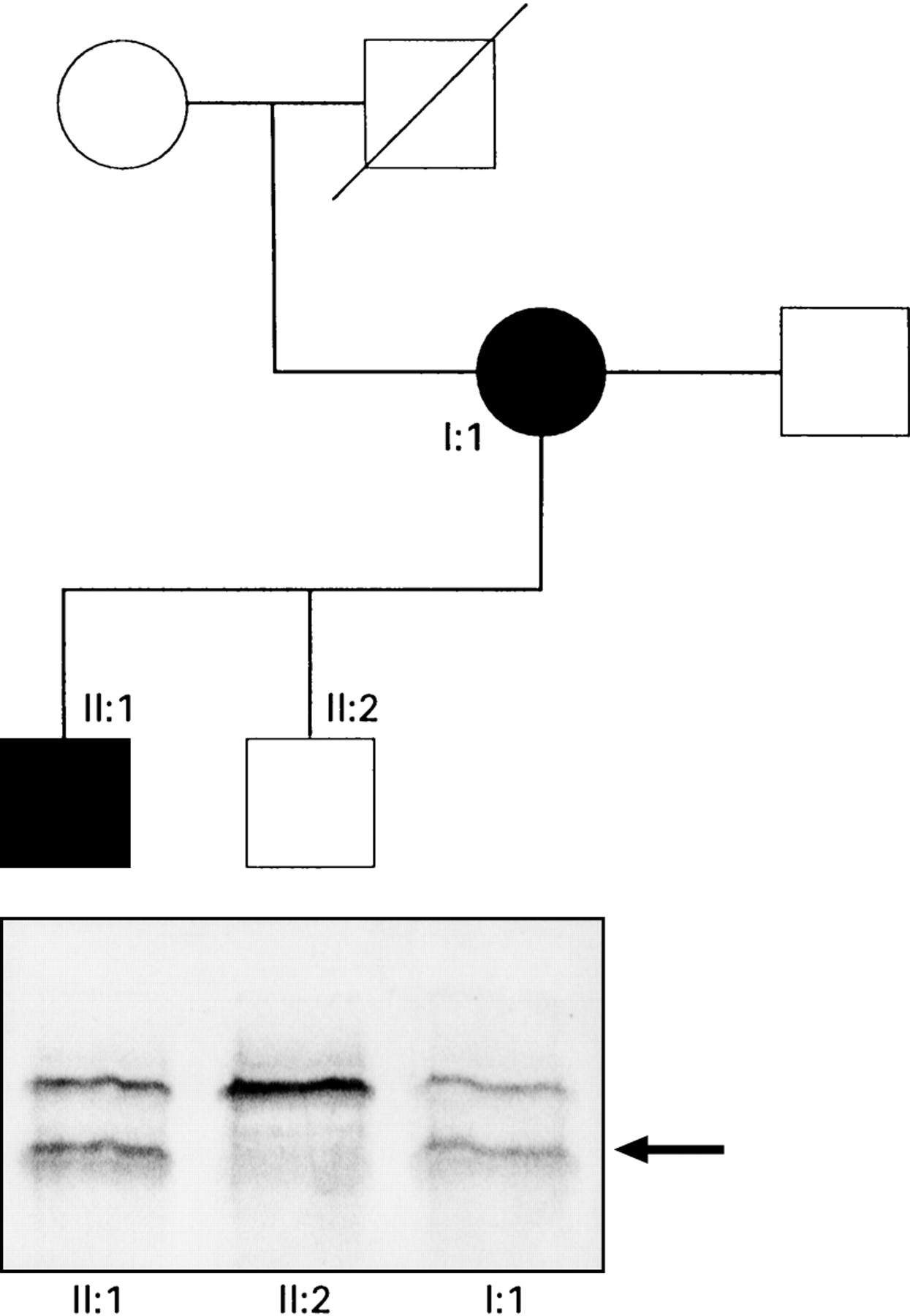
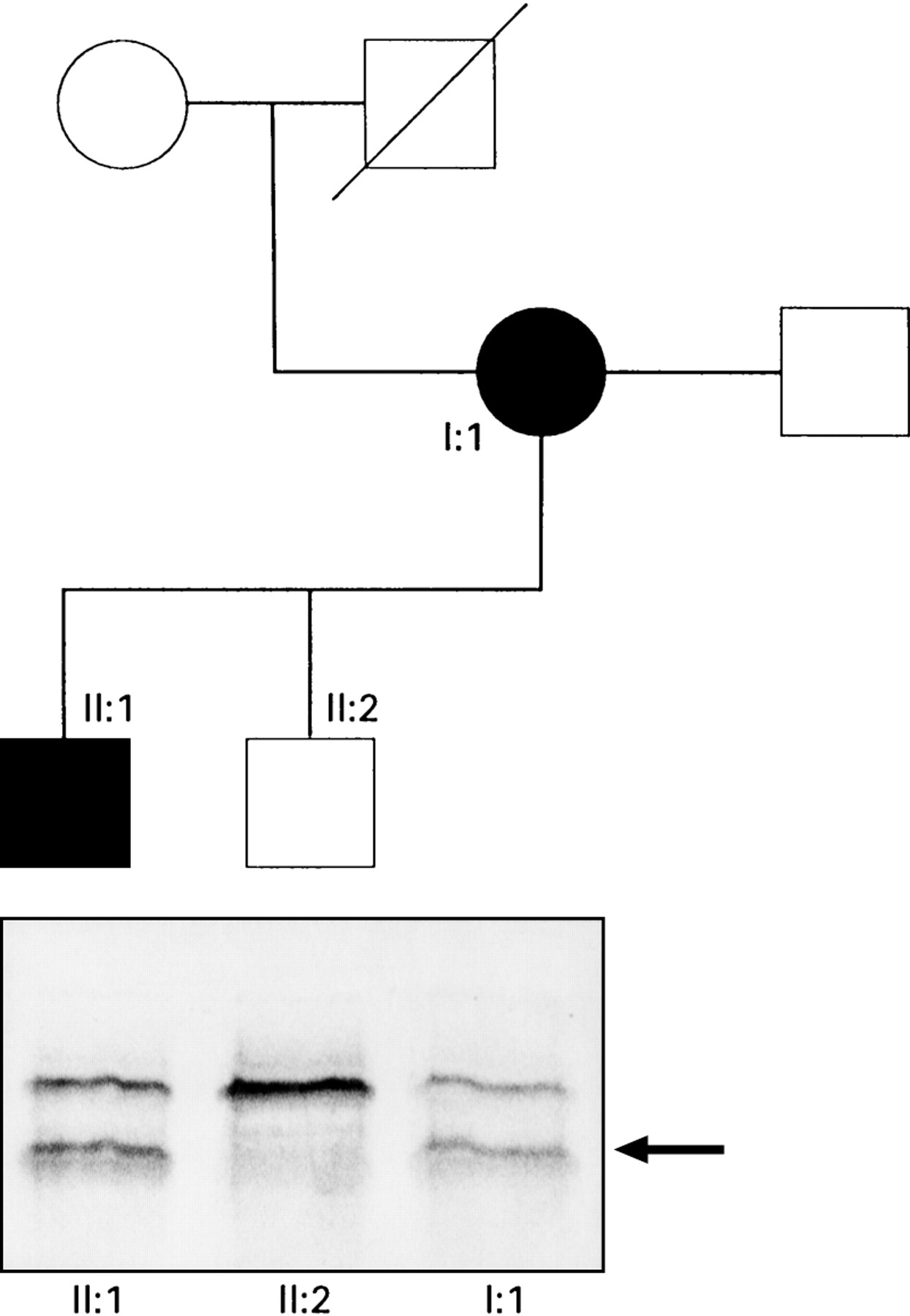
: Pedigree of the family. Circles represent females, squares males. Filled symbols represent individuals known to be affected. In the lower half of the figure are the results of the protein truncation test. An additional truncated protein product (arrowed) is present in samples from affected individuals following in vitro transcription/translation of exon 15 segment 3.
At age 38, upper endoscopy was performed because of dyspepsia. It showed a 3 cm sessile polyp of the pyloric canal which was removed endoscopically and histologically confirmed to be an adenoma. In addition there were multiple small polyps throughout the stomach and approximately 40 polyps in the first and second parts of the duodenum including a sessile villous adenoma occupying most of the first part. These lesions were subjected to extensive diathermy on multiple occasions. A radiological small bowel study showed no polyps in the remaining small intestine. At age 40 partial gastrectomy of the distal stomach was performed. Histology confirmed a 2.5 cm sessile tubulovillous adenoma of the pylorus and multiple fundic gland polyps. Moderate dysplasia was present in the large adenoma. At this time the patient had stage IV duodenal polyposis according to the Spigelman criteria.16 Despite continuing regular extensive diathermy of duodenal polyps she required a duodenotomy at age 45 to remove a very large sessile adenoma. Histology showed a specimen measuring 70×45×20 mm consisting entirely of tubulovillous adenoma with severe dysplasia. Her polyposis is considered particularly severe as both her stomach and duodenum are carpeted with polyps such that virtually no normal mucosa remains.
The proband’s two sons were screened. Individual II:2 (fig 1) had normal flexible sigmoidoscopies at ages 18 and 20 and a normal upper endoscopy at age 20. Genetic testing has shown that he does not carry the mutated APC gene. Individual II:1 had six colonic polyps removed at age 16 and was started on sulindac. Subsequent colonoscopy at age 18 was normal. Upper endosocpy at age 18 showed six duodenal adenomas and fundic gland polyps in the stomach. Upper endoscopy at age 21 showed more than 20 duodenal polyps, the largest of which was 5 mm in size. Histology showed tubular adenomas with mild dysplasia (Spigelman stage III). Colonoscopy at age 21, while not on sulindac, showed scattered polyps from caecum to rectum with less than 100 in total. Genetic testing shows him to have inherited the mutant APC gene. Examination for CHRPE was negative.
Protein truncation testing showed a truncated protein produced from exon 15 segment 3 (fig 1) in the proband (I:1) and individual II:1. These subjects showed positive heteroduplex bands in fragment 15–7. Sequencing of this fragment showed a 2 base pair deletion at codon 1520 thus changing the sequence from ...GAT GTG GAA... to ...GGT GGA A... This deletion causes a frameshift mutation which results in a stop codon 13 codons downstream. Figure 2 shows the location of this mutation within the APC gene. The locations of codons 1061 and 1309 which are hotspots for germline mutations are shown. Germline mutations after codon 1445 are associated with a high incidence of desmoid tumours.9 The region in which germline mutations are associated with CHRPE is shown.9 ,18 The lower portion of the figure shows the regions in which somatic mutations in tumours from FAP patients preferentially occur.17 ,19 ,20 The mutation in the family reported is at codon 1520 which is beyond the region of frequent mutation in colorectal tumours but within the regions mutated in duodenal and desmoid tumours. Mutations at or close to codon 1520 may selectively promote duodenal and desmoid tumorigenesis rather than profuse colonic polyposis.

{kind=link}
{kind=link}
: Schematic diagram of the central portion of the APC gene showing the region in which germline mutations known to cause FAP occur. Exons are labelled 8 to 15.
Discussion
This family has an unusual phenotype with sparse colonic polyposis but severe polyposis of the stomach and duodenum. The mutation falls within the region between codons 1445 and 1578 recently described by Caspari et al 9 and Davies et al. 10 In their studies only 8% and 15% respectively of mutations were found in this region; this is the only such mutation we have found in 18 unrelated families in whom APC mutations have been identified. This family corresponds to the phenotype described in that affected individuals do not express CHRPE.
Neither of these studies report the numbers of colonic polyps found in subjects with mutations in this region. Nagase et al 5 defined profuse polyposis as >5000 polyps and found it to be associated with mutations between codons 1250 and 1330. Mutations 5′ (proximal) to this caused sparse polyposis and the three cases with 3′ (distal) mutations at codons 1464 (two separate mutations) and 1597 had sparse polyposis. Subsequent studies confirmed these findings6 ,7 ,17 although in Western studies the total polyp count is lower; >2000 polyps usually represents profuse polyposis and <500 sparse polyposis. Our proband undoubtedly fits the sparse phenotype having had only 33 polyps at presentation at the age of 29. A recent study by Dobbie et al 11reported six families with a mutation 3′ of codon 1403 who had unpredictable and often severe phenotypes including “stomach polyps”. Scott et al 12 also reported a family with a 3′ mutation and a variable phenotype. These reports suggest that 3′ mutations may be associated with different phenotypes to those observed with more common mutations and are worthy of further study.
Somatic mutations occurring in colorectal neoplasms both in the sporadic and FAP settings are more closely clustered between codons 1280 and 1500 than the germline mutations causing FAP (fig2).18 Both mutations downstream of codon 1445 and mutations upstream of exon 9 are associated with sparse polyposis and lack of CHRPE19 which suggests that the malfunction of the APC protein which most efficiently causes colonic polyposis also causes CHRPE.
An earlier study did not show upper gastrointestinal polyps to be more common with any particular mutation but no patients with mutations between codons 1445 and 1578 were examined.17 Our family suggests that mutations in this region may result in very severe upper tract polyposis. Furthermore, somatic APC mutations in duodenal tumours preferentially occur between codons 1450 and 155620 and in desmoid tumours between codons 1399 and 158121 (fig 2). These correlations suggest that subtle differences in the function of mutant APC proteins may selectively promote tumorigenesis in different cell types.
An alternative explanation for the unusual phenotype observed in our family would be that a modifying gene is cosegregating with the disease. A modifier gene affects the expression of the mouse equivalent of FAP and an excellent candidate for this modifier gene is secretory phospholipase A2.22 ,23 One difficulty in postulating that a modifier gene is the cause of the unusual phenotype in our family is that such a gene would need to reduce colonic polyposis while increasing duodenal polyposis. Secretory phospholipase A2 is thought to act by increasing arachidonic acid production and thus cell signalling via prostaglandins and leukotrienes to reduce polyp formation. To explain the phenotype in the present family, it would need to have widely differing activities in the colon and duodenum.
In summary, we have described a family with FAP due to a mutation in APC codon 1520 who have sparse colonic polyposis but severe upper gastrointestinal polyposis. The small number of cases in this family limits interpretation of the data and further cases need to be studied to determine whether this unusual phenotype is due to the location of the mutation or a modifier gene which has differing effects in the duodenum and the colon. From a practical point of view this family demonstrates the need for upper tract surveillance even in FAP individuals with mild colonic disease.
Acknowledgments
This study was supported by the Queensland Cancer Fund and the Department of Pathology, Royal Brisbane Hospital.