Article Text

Abstract
Background—Recent reports have described a modulating influence of nitric oxide (NO) on intestinal mucosal permeability and have implicated a role for mast cells in this NO mediated process.
Aims—To assess further the contribution of mast cells to the mucosal permeability changes elicited by the NO synthase (NOS) inhibitor NG-nitro-l-arginine methylester (l-NAME), using mast cell deficient (W/WV) and mast cell replete mice (+/+).
Methods—Chromium-51 EDTA clearance (from blood to jejunal lumen), jejunal NOS and myeloperoxidase (MPO) activities, and plasma nitrate/nitrite levels were monitored.
Results—The increased EDTA clearance elicited by intraluminal l-NAME in W/WV mice (4.4-fold) was significantly greater than the response observed in control (+/+) mice (1.8-fold). The exacerbated response in W/Wv mice was greatly attenuated by pretreatment with either dexamethasone (1.3-fold) or the selective inducible NOS inhibitor, aminoguanidine (1.4-fold), and partially attenuated by the mast cell stabiliser, lodoxamide (2.9-fold). Jejunal inducible NOS activity was significantly higher in W/WV than in +/+ mice, while jejunal MPO was lower in W/WV mice than in +/+ mice, suggesting that the higher inducible NOS in W/WV does not result from the recruitment of inflammatory cells into the gut. The higher inducible NOS activity in the jejunum of W/WV was significantly reduced by dexamethasone treatment.
Conclusions—Our results suggest that mast cells normally serve to inhibit inducible NOS activity tonically in the gut and that inhibitors of NOS elicit a larger permeability response when this tonic inhibitory influence is released by mast cell depletion.
- aminoguanidine
- c-kit
- dexamethasone
- epithelium
- neutrophils
Statistics from Altmetric.com
There is a growing body of evidence that implicates nitric oxide (NO) as a modulator of the function of different cells that are normally found within the gastrointestinal mucosa, including epithelial cells,1 mast cells,2 endothelial cells, and leucocytes.3-5 It appears that NO also contributes to the interactions between these different cell populations in certain pathological conditions, such as ischaemia reperfusion.6 ,7 NO mediated cell-cell interactions have been described between mast cells, leucocytes, and enterocytes in intestinal segments treated with NO synthase inhibitors, such as NG-nitro-l-arginine methylester (l-NAME) or NG-monomethyl-l-arginine (l-NMMA).8 These inhibitors elicit the degranulation of mast cells, recruitment of leucocytes, and increased mucosal permeability to chromium-51 EDTA, responses that are prevented by simultaneous treatment with either NO donating agents orl-arginine.5 ,9 Mast cell stabilising agents such as doxantrazole and lodoxamide, as well as histamine H1 antagonists, also appear to be effective in blunting the increased mucosal permeability associated with NO synthase inhibition.10 Hence, it has been proposed that mast cell derived histamine, which is released from degranulated mast cells that are deprived of NO, mediates the changes in paracellular permeability in the gut after NO synthase inhibition.
The linkage between diminished intestinal NO production, mast cell degranulation, and increased mucosal permeability has been invoked to explain the mucosal barrier dysfunction observed after ischaemia reperfusion.8 This NO dependent mechanism may also account for the increased intestinal mucosal permeability observed in inflammatory conditions that are associated with an enhanced superoxide mediated inactivation of NO. A limitation of this novel mechanism is that the implication of mast cells as a modulator of the increased mucosal permeability elicited by NO synthase inhibition is largely derived from experiments utilising agents that are considered to act exclusively through mast stabilisation. Since there is published evidence that challenges the specificity of mast cell stabilisers such as ketotifen,11 an alternate approach to testing the linkage between mast cells and intestinal mucosal permeability is warranted. The objective of this study was therefore to assess further the contribution of mast cells to the increased mucosal permeability elicited by NO synthase inhibition using mice that are genetically deficient in mast cells.
Methods
ANIMALS
Male (20–30 g) mast cell deficient mice (WBB6F1/JW/WV; W/WV) (n=62) and their age matched controls (WBB6F1/J+/+; +/+) (n=24) were obtained from Jackson Laboratories (Bar Harbor, Maine). Male ICR mice (n=6), another strain of control mice, were obtained from Harlan Laboratories (Frederick, Maryland). Only occasional mast cells (less than 1% of the number in congenic +/+ mice) are observed in the skin of adult W/WVmice, and no mast cells have been noted in multiple other tissues of W/WV mice.12 ,13 Similarly, we have been unable to find mast cells in the mesentery of 12 week old W/WV mice.
MUCOSAL PERMEABILITY
The blood to lumen clearance of 51Cr labelled EDTA, modified for mice from the conventional method previously applied in rats and cats,9 ,10 was measured in the jejunum of W/WV, +/+, and ICR mice. Animals were fasted for 24 hours prior to surgery. Anaesthesia was induced by intramuscular injection of ketamine (150 mg/kg) and xylazine (10 mg/kg), with supplemental doses of subcutaneous ketamine as needed. The right carotid artery and left jugular vein were cannulated. Systemic arterial pressure was measured with a Statham (Oxnard, California) P23A pressure transducer and recorded with a Grass polygraph DC driver amplifier (Grass Instrument Co., Quincy, Massachusetts). A laparotomy was performed, and a loop of jejunum (4–5 cm) was isolated; blood vessels were left intact. Inflow and outflow tubes were secured on each end of the jejunal segment, and warm phosphate (10 mM) buffered saline (pH 7.4) was perfused through the segment at a rate of 0.5 ml/min. The intestinal segment and abdominal contents were kept moist with saline soaked gauze and covered with a clear plastic sheet to minimise evaporation and tissue dehydration. Body temperature was monitored and maintained at 36.5–37.5°C by a heat lamp.
Since 51Cr-EDTA is rapidly excreted into urine following intravenous injection (unless the kidneys are ligated), we continuously infused 35 μCi (11.7 g/ml; 0.2 μCi/min) of the tracer into the jugular vein. This regimen of 51Cr-EDTA administration was determined in initial experiments in which the bolus and infusion levels were varied until one was found to yield a constant plasma level. The appropriateness of the chosen bolus dose and infusion rate of 51Cr-EDTA is supported by the stable clearance values observed over 80 minutes of luminal perfusion (fig 1). Thirty minutes were allowed for tissue equilibration of 51Cr-EDTA before collecting samples of luminal perfusate (every 10 minutes). Plasma samples (25 μl) were obtained immediately prior to collection of the luminal perfusate (time 0) and at the end of the experiment (at 80 minutes). Plasma EDTA level for each time point was calculated from an assumed linear curve obtained by 51Cr count of the two plasma samples. 51Cr activity in plasma and luminal perfusate was counted in a 14800 Wizard 3 gamma counter (Wallac, Turku, Finland), with automatic correction for background activity. At the end of the experiment the jejunal loop was removed, rinsed, and weighed. The plasma to lumen clearance of 51Cr-EDTA was calculated as:

: (A) Time course of mucosal permeability changes in untreated controls (+/+) (n=5), and elicited by l-NAME in +/+ (n=6), and mast cell deficient (W/WV) mice (n=6). (B)l-NAME induced mucosal permeability changes expressed as a ratio of the maximum clearance to baseline value (10–20 minute clearance value) in the following groups: +/+, W/WV, W/WV pretreated with lodoxamide (Lod) (n=5), W/WV pretreated with dexamethasone (Dex) (n=5), and W/WV pretreated with aminoguanidine (AG) (n=6) and ICR mice (n=6). *p<0.05 relative to respective baseline value, †p<0.001 relative to l-NAME-treated +/+, *p<0.05, ‡p<0.001 relative to the peak clearance response of W/WV.
Clearance (ml/min × 100g) = (CPM, perfusate) × (perfusion rate) × 100 (CPM, plasma) × (wt of intestinal segment)where CPM is counts per minute per ml of perfusate or plasma, and wt is weight in grams of the jejunal segment. The theoretical basis and technical limitations of the 51Cr-EDTA clearance technique for measurement of intestinal mucosal permeability have been addressed elsewhere.14
Stable baseline clearances of 51Cr-EDTA in murine intestine were noted for as long as 80 minutes (fig 1A). l-NAME(1 mM, Sigma, St Louis, Missouri) was perfused into the intestinal lumen for 60 minutes, which was preceded by a 20 minute period of control perfusion without l-NAME (baseline measurements). Preparations with a baseline clearance greater than 0.2 ml/min/100 g were discarded. Lower concentrations of l-NAME(0.1–0.5 mM) in the gut lumen did not elicit an increase in51Cr- EDTA clearance in control mice (unpublished data), as shown in previous studies.8 ,9 It has been reported that concentrations of l-NAME as high as 1 mM do not increase the permeability of monolayers of rat intestinal epithelial cells.10
In another series of experiments, the mast cell stabiliser lodoxamide (15 mg/kg), was administered intravenously as a bolus at 15 minutes before the start of the experiment, in W/WVmice.10 Other animals (W/WV) were pretreated with either dexamethasone (1 mg/kg intraperitoneally, Sigma, St Louis, Missouri) 24 hours before the experiment, or with the selective inducible nitric oxide synthase (iNOS) inhibitor, aminoguanidine.15-17 Aminoguanidine was administered in the drinking water (18.5 mg/l) for three days before the experiment, a regimen which has been shown to blunt the increased plasma nitrate and nitrite levels normally elicited by peptidoglycan/polysaccharide in rats.18
NOS AND MYELOPEROXIDASE (MPO) ACTIVITIES
In additional groups of fasted (24 hours) W/WV and +/+ mice, samples of jejunum were harvested and quickly frozen for later measurement of NOS or MPO activity. Jejunal NOS activity was determined using the method of Kurose et al.5 MPO activity was determined using the method of Grisham et al 18 in jejunal samples of untreated W/WV and +/+ mice.
PLASMA NITRITE AND NITRATE LEVELS
Plasma nitrite and nitrate levels in W/WV and +/+ mice were measured using the method of Grisham et al.19 Animals were fasted for 24 hours, and a blood sample was collected from the carotid artery. Plasma nitrate and nitrite levels were also measured in dexamethasone or aminoguanidine pretreated W/WV mice under basal conditions.
STATISTICS
The data were analysed using an analysis of variance with Scheffé’s (post hoc) test. When only two groups were compared, the paired or unpaired Student’s t test was used where appropriate. All values are reported as means (SEM). Statistical significance was set at p<0.05.
Results
MUCOSAL PERMEABILITY
The blood to lumen clearance of 51Cr labelled EDTA was significantly increased by the infusion of l-NAME in W/WV mice, but not in +/+ mice (fig 1A). Figure 1B shows the peak changes in l-NAME induced EDTA clearance (normalised to its baseline value) in the different groups of animals. Although l-NAME caused a small increase in EDTA clearance (1.8× baseline) in control (+/+) mice, a more substantial increase (4.4× baseline) was elicited by l-NAME in W/WV mice. This l-NAME induced increase in the mucosal permeability observed in W/WV mice was slightly attenuated by lodoxamide (2.9× baseline) and greatly attenuated by pretreatment with either dexamethasone or aminoguanidine. The magnitude of the increase in EDTA clearance elicited by l-NAME in another strain of mast cell replete mice (ICR) was similar to that observed in +/+ controls.
Systemic arterial pressure was unaffected by infusion ofl-NAME in all groups studied.
JEJUNAL NOS ACTIVITY
Total NOS activity under baseline conditions was significantly higher in the jejunum of W/WV mice than in (+/+) control mice (fig 2). This difference between W/WV and +/+ mice was more evident for iNOS activity. The elevated iNOS in W/WVmice was significantly attenuated by pretreatment with dexamethasone, but not by aminoguanidine pretreatment. The activity of constitutive NOS (cNOS) in mouse jejunum did not differ between the various experimental groups.
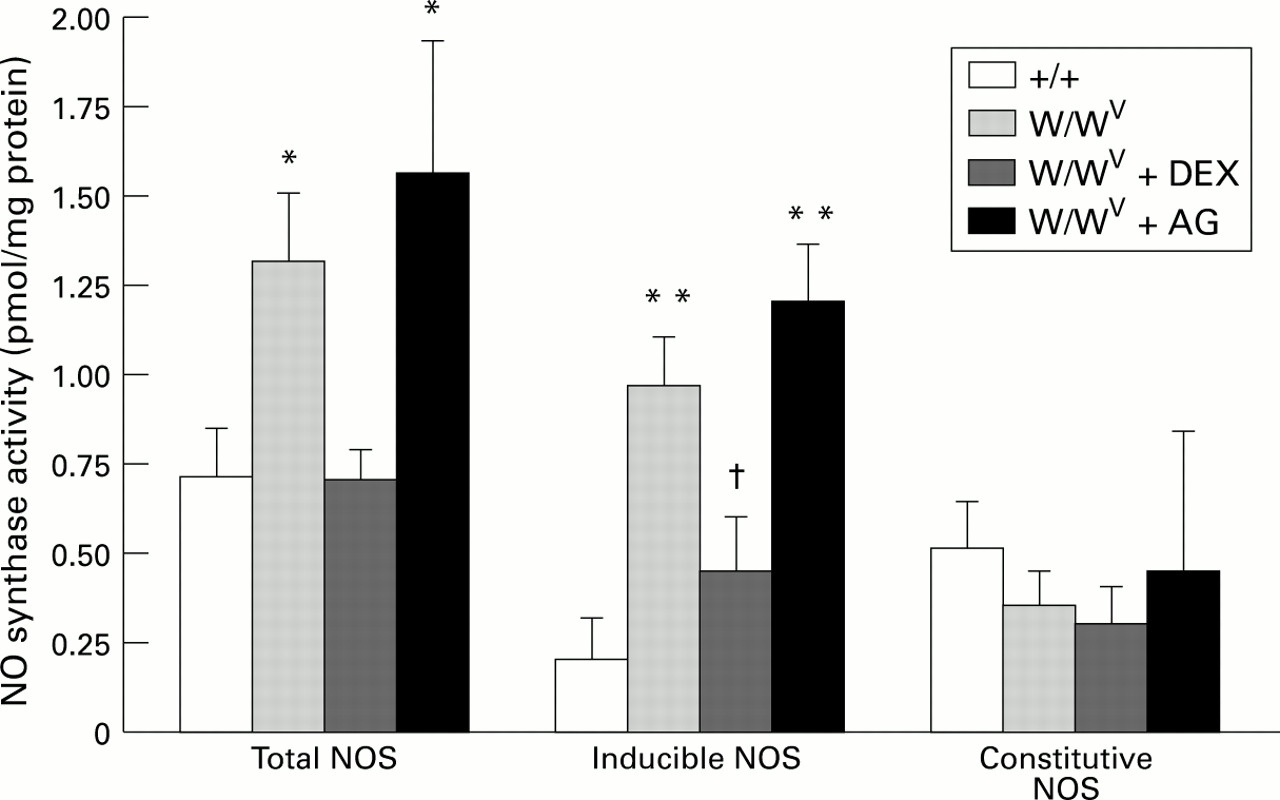
: Nitric oxide synthase (NOS) activity in control (+/+) mice (n=7), mast cell deficient (W/WV) mice (n=8), W/WV mice pretreated with dexamethasone (DEX) (n=8), and W/WV mice pretreated with aminoguanidine (AG) (n=6). *p<0.05, **p<0.001 relative to +/+, †p<0.05 relative to W/WV.
JEJUNAL MPO ACTIVITY
Tissue MPO activity in jejunum of W/WV mice (6.52 (0.38) U/g) was significantly lower (p<0.001) than that of +/+ mice (11.31 (0.53) U/g).
PLASMA NITRATE AND NITRITE
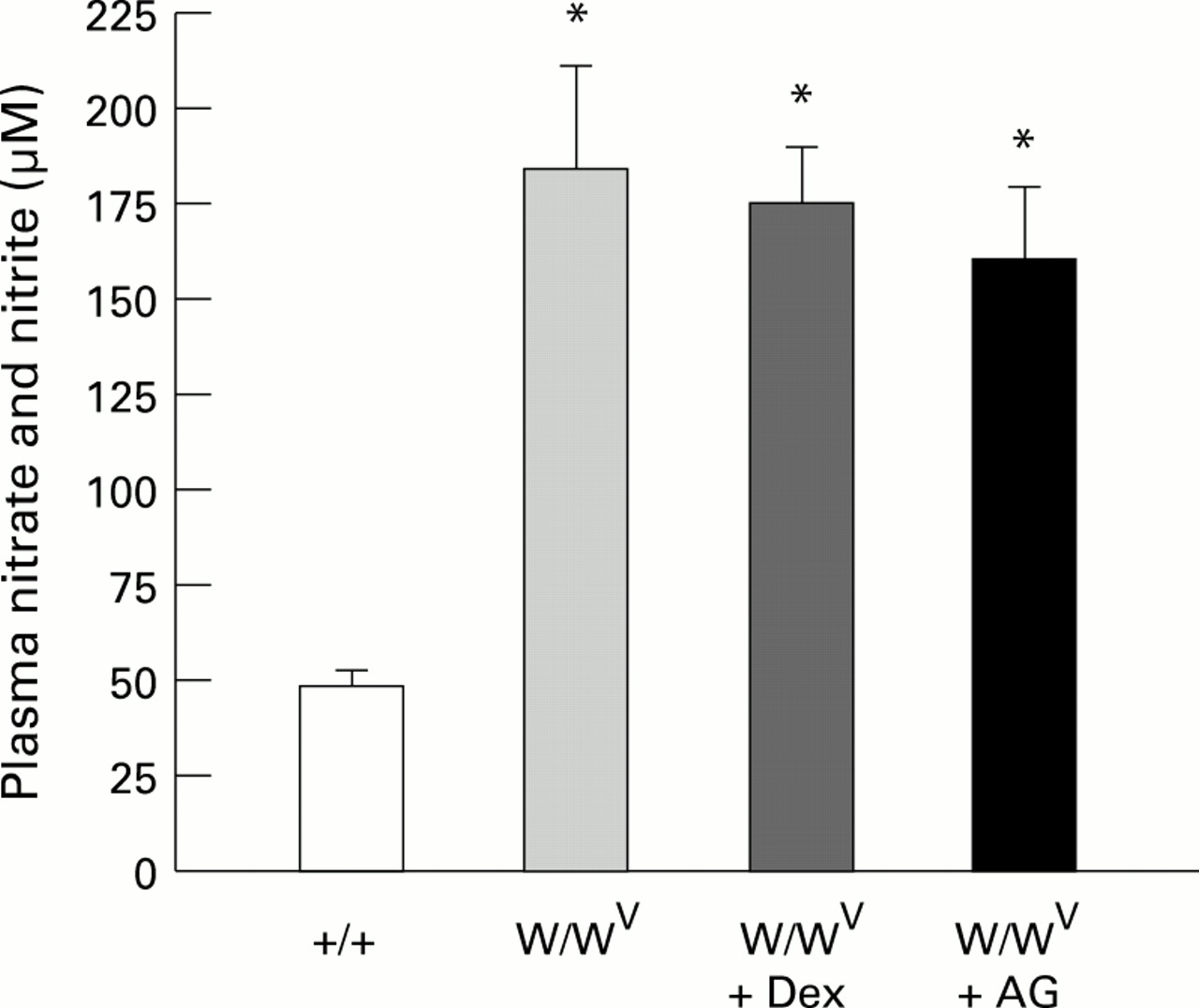
The plasma level of nitrite/nitrate (NO2/NO3) in W/WV mice was significantly higher than that measured in control (+/+) mice. The higher levels of plasma NO2/NO3 in W/WV mice were not significantly attenuated by pretreatment with either dexamethasone or aminoguanidine (fig 3). The absence of a change in plasma NO2/NO3 in the face of a profound reduction in iNOS activity after dexamethasone treatment may reflect a differential sensitivity of various organs to dexamethasone. We measured NOS activity in the jejunum but not in other tissues, which may contribute more significantly to plasma NO2/NO3 in mast cell deficient mice. This possibility, however, is not supported by previously published work which demonstrates that dexamethasone (1 mg/kg) inhibits iNOS activity to a comparable extent in different tissues of the rat after endotoxin treatment.20 Hence, an alternative explanation for our findings of a sustained elevation in plasma NO2/NO3 and accompanying reduction in iNOS after dexamethasone or aminoguanidine treatment is that the increased plasma NO2/NO3 level in mast cell deficient mice is not related to the elevated jejunal iNOS activity.
{kind=link}
{kind=link}
{kind=link}
: Plasma levels of nitrite and nitrate in control (+/+) (n=6), mast cell deficient (W/WV) (n=6), and W/WV mice pretreated with either dexamethasone (Dex) (n=6) or aminoguanidine (AG) (n=6). *p<0.01 relative to +/+.
Discussion
There is a growing body of evidence that implicates nitric oxide as a modulator of the restrictive properties of the paracellular pathway between intestinal epithelial cells.8 Using blood to lumen clearance of 51Cr-EDTA as a measure of intestinal mucosal permeability, Kubes and associates9 ,10 have demonstrated that intravascular administration of NO synthase inhibitors such as l-NAME elicit large (fivefold to sixfold) increases in mucosal permeability of both cat and rat intestine. They also showed that this effect of l-NAME was completely reversed by simultaneous administration of either nitroprusside or l-arginine, but could not be mimicked by administration of the inactive enantiomer, d-NAME. Based on these observations, the authors proposed that nitric oxide produced in the gut mucosa exerts a tonic inhibitory influence on the passive permeation of solutes into the bowel lumen, presumably through a direct (or indirect) action of NO on epithelial junctions. Support for an indirect action is provided by experiments demonstrating thatl-NAME does not increase 51Cr-EDTA clearance across monolayers of cultured rat intestinal epithelial cells.10
Our observation that perfusion of the lumen of mouse jejunum withl-NAME elicits an increased blood to lumen clearance of51Cr- EDTA provides additional support for the view that acute inhibition of NO production leads to an increased mucosal permeability. However, the magnitude of the l-NAME induced increase in 51Cr-EDTA clearance elicited in the jejunum of control mice, that is, +/+ (1.8-fold) and ICR (2.5-fold), is significantly lower than that previously observed in rat and cat intestine.9 ,10 Although a definitive explanation for the different responses to l-NAME is not available, two possibilities warrant consideration. Firstly, it appears likely that the luminal route of l-NAME administration used in our study achieves a lower concentration of the NO synthase inhibitor in the mucosa than that achieved following intravascular (intravenous or intra-arterial) administration. Another possibility is that mice respond less vigorously to NO synthase inhibition (or have a lower activity of the enzyme) than other species (rat or cat). This possibility seems less plausible since we have noted in a few rat intestine preparations that luminal l-NAME elicits an increased 51Cr-EDTA clearance comparable to that observed in mouse intestine (data not shown).
There is published evidence that implicates mast cells in the enhanced epithelial barrier permeability associated with inhibition of NO synthase.8 ,10 Kanwar et al 10have shown that mast cell stabilisers or histamine H1receptor antagonists greatly attenuate the increased mucosal permeability normally elicited by l-NAME, and that chronic dexamethasone treatment, which depletes mucosal mast cells, also prevents the l-NAME induced rise in mucosal permeability. These observations suggest that mast cell derived histamine mediates the increased mucosal permeability caused by l-NAME. The assumption that NO synthase inhibition promotes mast cell degranulation is supported by the observation that mast cell protease II activity in plasma is increased in animals receiving l-NAME, compared with control plasma. Additional support for a linkage between mast cells and NO production is provided by intravital microscopic studies of rat mesentery, in which l-NAME5 or ischaemia reperfusion7 induced mast cell degranulation is blunted by coadministration of NO donating agents (for example, spermine NO).
The results of the present study are inconsistent with the prevailing view that mast cell products (for example, histamine) are responsible for the increased intestinal mucosal permeability that is associated with inhibition of NO production. Surprisingly, we found that l-NAME elicits a substantially larger increase in51Cr-EDTA clearance in jejunal segments of mast cell deficient mice (W/WV) compared with their mast cell replete counterparts (+/+ mice). This finding contrasts the profound protective action of mast cell stabilisers, such as lodoxamide and doxantrazole, on l-NAME induced mucosal permeability changes observed in rat small intestine.10 Our findings in mast cell deficient mice would argue in favour of a role for mast cells in producing factors that protect the intestinal mucosa against the permeability increasing effects of NO synthase inhibition.
While a definitive explanation is not readily available for the discrepancy between our findings in mast cell deficient mice and previously published work on NO synthase inhibition in normal intestine treated with mast cell stabilisers, some mechanistic insights may be afforded by the responses of other variables measured in our experiments. The clearance response elicited by l-NAME in mast cell deficient mice was slightly but significantly attenuated by pretreatment with the widely used mast cell stabiliser lodoxamide, suggesting that this agent may exert an action on the mucosal barrier that is independent of mast cells. While such non-specific actions of mast cell stabilisers could account, at least in part, for our contradictory results, adaptive responses related to the chronic absence of mast cells in the intestinal mucosa may contribute more significantly to the larger permeability response observed in the mutant mice.
We noted that jejunal MPO (an index of tissue granulocyte count) was reduced, while jejunal iNOS (but not cNOS) activity was greatly elevated in mast cell deficient mice, compared with their mast cell replete counterparts. The profound increase in jejunal iNOS activity was accompanied by a large increase in plasma nitrate/nitrite concentration, an index of total body NO production. The relatively low jejunal MPO activity in W/WV mice suggests that an accumulation of activated granulocytes is an unlikely explanation for the greater 51Cr-EDTA clearance response in these mutants. However, the greatly elevated iNOS activity (and plasma NO2 –/NO3 –) in W/WV mice may relate more directly to the exaggerated51Cr-EDTA clearance response noted in these animals.
Our observations that plasma NO2/NO3 is greatly elevated and that jejunal iNOS activity is increased fivefold in W/WV mice suggests that chronic mast cell deficiency is associated with an accelerated rate of NO production in the gut (and possibly other tissues). Whether or not this enhanced NO generation is causally related to the exaggerated mucosal permeability response tol-NAME in mast cell deficient mice remains unclear. However, some support for this possibility is provided by the observation that pretreatment of W/WV mice with dexamethasone, which abolished the rise in iNOS normally associated with mast cell depletion, resulted in an attenuated51Cr-EDTA clearance response to l-NAME. Similarly, treatment with aminoguanidine, which is a more selective competitive inhibitor of iNOS15-17 (l-NAME is more specific for cNOS),21 also blunts the rise in mucosal permeability that is elicited by l-NAME in W/WVmice. Overall, these findings suggest that mast cells normally serve to inhibit iNOS activity tonically in the gut and that l-NAMEelicits a larger permeability response when this tonic inhibitory influence is released by mast cell depletion. Nonetheless, it remains unclear how iNOS inhibitors such as aminoguanidine and dexamethasone act to protect against the actions of another inhibitor of NOS—that is, l-NAME. It is conceivable that the beneficial actions of aminoguanidine and dexamethasone are unrelated to iNOS inhibition. Indeed, it has been shown that aminoguanidine has other biological actions, including inhibition of diamine oxidase.22 ,23Alternatively, a negative feedback to iNOS by NO24 may be released by selective inhibition of cNOS with a relatively lower dose of l-NAME,21 with the elevated NO levels mediating the rise in mucosal permeability.
The induction of iNOS in mast cell deficient mice is not likely to result from the recruitment of inflammatory cells into the jejunum inasmuch as tissue MPO activity was significantly lower in W/WV than in control (+/+) mice. Another consideration relates to the mutations at the W/c-kit locus25 ,26 in W/WV mice, which results either in an absence of c-kit tyrosine kinase receptor on cell surfaces or in the expression of receptors that are notably deficient in tyrosine kinase activity.26-28 Thus, it is conceivable that inactivation of the receptor tyrosine kinase encoded by c-kit could induce iNOS expression. However, this possibility appears unlikely in view of recent reports demonstrating that inhibition of tyrosine kinase attenuates the induction of iNOS expression, indicating that tyrosine kinase is necessary for iNOS expression.29 ,30 A more tenable explanation for the increased jejunal iNOS activity in W/WV mice is that mast cells produce factors that suppress the gene for iNOS. Indeed, Dileepan et al 31showed that mast cell granules inhibited lipopolysaccharide (LPS) or interferon γ induced nitric oxide production in peritoneal macrophages, suggesting an inhibitory effect of mast cell products on iNOS expression. Furthermore, it has been shown that mast cell derived factors such as interleukin (IL) 4,32 IL-13, or transforming growth factor β33 inhibit the expression of iNOS.34 ,35
In summary, the findings of this study, coupled with published reports, suggest that the modulatory role of mast cells in mediating the increased mucosal permeability that is elicited by l-NAMEdiffers between conditions of acute versus chronic suppression of mast cell function. Our results support the view that c-kit (and presumably mast cells) play an important role in maintaining epithelial barrier integrity in the small intestine. However, the possibility cannot be excluded that other cells mediate this process, since the depletion of c-kit receptor tyrosine kinase affects different cell populations, such as interstitial cells of Cajal, to regulate gastrointestinal motility,36 as well as haemopoietic cells.
Acknowledgments
Supported by a grant from the National Institutes of Health (P01 DK43785).