Article Text

Abstract
Background—Massive liver necrosis, characteristic of acute liver failure, may affect hepatosplanchnic haemodynamics, and contribute to the alterations in renal haemodynamics and function.
Aims—To investigate the relation between hepatosplanchnic haemodynamics, including portal systemic shunting, and renal blood flow and function in rats with acute liver failure.
Methods—Liver failure was induced in male Wistar rats by intraperitoneal injection of 1.1 g/kg ofd(+)-galactosamine hydrochloride. The parameters assessed included: systemic, hepatosplanchnic, and renal blood flow (57Co microsphere method); portal-systemic shunting and intrarenal shunting (consecutive intrasplenic, intraportal, or renal arterial injections of 99mTc methylene diphosphonate and99mTc albumin microspheres); arterial blood pressure and portal pressure; renal function; and liver function (liver function tests and 14C aminopyrine breath test).
Results—Progressive liver dysfuntion was accompanied by the development of a hyperdynamic circulation, a highly significant decrease in renal blood flow and function, and an increase in intrarenal shunting 36, 42, and 48 hours after administration of d-galactosamine. The alterations in renal blood flow and function were accompanied by significant increases in portal pressure, portal venous inflow, and intrahepatic portal systemic shunting in galactosamine treated rats compared with controls. There was a significant correlation between changes in renal blood flow and changes in portal pressure, intrahepatic portal systemic shunting, and deterioration in liver function (r=0.8, p<0.0001).
Conclusions—The results of this study suggest that both increased intrahepatic portal systemic shunting and hepatocyte impairment may contribute to alterations in renal haemodynamics and function.
- liver failure
- hepatosplanchnic
- systemic
- renal
- haemodynamics
Statistics from Altmetric.com
Acute liver failure, characterised by severe hepatocyte injury or massive hepatic necrosis is accompanied by oliguric renal failure in approximately 50% of patients.1 Functional renal failure (hepatorenal syndrome) is usually present but acute tubular necrosis has also been reported.2 These changes in kidney function are accompanied by a reduction in renal blood flow and effective renal plasma flow secondary to decreased perfusion pressure, intrarenal vasoconstriction, increased intrarenal shunting, or a combination of these effects.3-6 Furthermore, acute liver failure is characterised by profound alterations in systemic haemodynamics including hypotension, a decrease in splanchnic vascular resistance, pooling of blood in the splanchnic circulation, a compensatory increase in cardiac output, and a supply dependent oxygen consumption indicative of an underlying tissue oxygen debt.6-10 The circulatory changes characteristic of acute liver failure resemble those observed in cirrhosis in some respects but differ in others. Thus, in cirrhosis and portal hypertension the characteristic hyperdynamic circulation and alterations in renal haemodynamics are thought to result from increased circulating levels of gut derived vasoactive agents which are normally removed by the liver, entering the systemic circulation as a consequence of impaired hepatocyte function and portal-systemic shunting (PSS).11-13 In contrast, in acute liver failure where there is no underlying chronic disease, the duration of the condition is considered to be too short for the development of significant PSS. Therefore, in acute liver failure, the increased systemic levels of the gut derived substances believed to be responsible for the changes in systemic and renal haemodynamics are thought to result primarily from the severe impairment of hepatic function. However, massive liver necrosis characteristic of acute liver failure, particularly the hyperacute form of the syndrome, may grossly affect hepatosplanchnic haemodynamics, and consequently contribute to the alterations in renal haemodynamics and function. As there is a paucity of data on the relation between hepatosplanchnic haemodynamics and PSS, and renal function and blood flow in acute liver failure we have carried out such a study in rats.
Materials and methods
EXPERIMENTAL DESIGN
Forty eight male Wistar rats (body weight 250–300 g), obtained from Harlan UK Ltd (Shaw’s Farm, Blackthorn, Bicester, Oxon) received an intraperitoneal injection of 1.1 g/kg ofd(+)-galactosamine hydrochloride (Sigma Chemical Corporation, St Louis, Missouri, USA) as a 100 mg/ml solution (pH 6.8) in normal saline.14 A control group of 48 rats received the same volume of normal saline. All animals were allowed access to a pelleted laboratory diet (CRM Labsure Animal Diets, K&K Greef Ltd, Croydon) ad libitum throughout the study period. Control animals were allowed access to water, and galactosamine treated animals to 10% glucose solution ad libitum to maintain blood glucose levels.
The rats were randomly allocated to four groups of 12 animals each for investigation at 24, 36, 42, and 48 hours after administration of either galactosamine or saline.
HAEMODYNAMIC MEASUREMENTS
At the time of study, the rats were anaesthetised by an intraperitoneal injection of pentobarbitone sodium 50 mg/kg (Sagatal, May & Baker Ltd). Through a midline incision the portal vein was exposed, cannulated via an ileocolic branch, and the cannula advanced to just below the porta hepatis. The midline laparotomy was closed in two layers after placement of the portal venous cannula to reduce evaporative loss and conserve body heat. The spleen was exposed via a small left lateral incision for the intrasplenic injection of radionuclides for the measurement of total PSS. The left and right femoral arteries and the left femoral vein were cannulated with 2FG Portex (Laboratoire Portex SA, France) cannulae. Arterial blood pressure and portal venous pressure were continuously monitored throughout the study by connecting the left femoral artery and portal venous cannulae via isolated pressure tranducers (Medex Ltd) and amplifier to a two channel recorder (MX2, Lectromed UK Ltd). Finally the right common carotid artery was exposed through a cervical incision and a 2FG polyethylene cannula was introduced into the carotid artery via an arteriotomy. The cannula was screened into the left ventricle using an image intensifier (Seimens Siremobile, Seimens Ltd, Sunbury-on-Thames, UK) so that its tip was approximately 1 mm below the aortic valve. Adequate expulsion of a small bolus of injectate by the left ventricle was confirmed by injecting 0.1 ml of the contrast medium, sodium meglumine loxaglate (320 mg/ml; May & Baker, Dagenham), via the catheter.
Portal-systemic shunting was determined by consecutive intrasplenic (or intraportal) injections of technetium-99m methylene diphosphonate (MDP) and 99mTc albumin microspheres as previously described.15 In brief, when the animals were haemodynamically stable, a straight bore sodium iodide scintillation counter linked to a scalemeter and computer (Spherex monitoring system, Pharmacia, Sweden) was placed over the rat’s thorax and shielded from the liver by a lead screen. The increase in count rate in the lung field of interest was calculated as the difference between the plateau level before each injection of the isotope and the baseline level. The baseline level was determined from the mean of the count rate during the 30 seconds immediately before the start of each injection. The plateau level was calculated as the mean count rate during 60 seconds (the average number of counts per second for six consecutive 10 second periods during the stable phase) within three minutes from the time of each injection of the isotope. The count rate was corrected for the dose of radioisotope in each injection to obtain comparable values of the count rate per unit of radioactivity injected. The decay of 99mTc (t1/2 = 6 hours) and the “dead time” of the detector were corrected for by the computer software to obtain a linear response up to a maximum count rate of 60 000 cps. Intrahepatic PSS was measured by injecting 20 μl (100 Mbq/ml) of 99mTc MDP followed by a 20 μl saline chaser via the portal venous cannula. As 99mTc MDP is not retained within the liver, the majority passes to the lungs and is registered graphically as the 100% passing fraction. The99mTc MDP therefore acts as a reference injection. Following injection of 99mTc MDP, 20 μl (100 MBq/ml) of vortex mixed 99mTc albumin microspheres (mean diameter 20 μm; Sferotonic-S, Sorin Biomedica, Italy), were administered via the portal venous cannula, again followed by a 20 μl saline chaser. Previous preliminary studies indicated that this volume of chaser ensures that all the isotope is washed out of the cannula into the portal vein. Unlike 99mTc MDP, the 99mTc albumin microspheres are trapped within the liver sinusoids and only those passing through the liver via the PSS appear in the lung field of interest. The percentage of the passing fraction of 99mTc albumin microspheres is expressed as a passing fraction of the reference injection of 99mTc MDP. Total PSS was determined by consecutive intrasplenic injections of 99mTc MDP and99mTc albumin microspheres. Extrahepatic PSS was calculated by subtracting the intrahepatic PSS from the total PSS. Previous studies have indicated an excellent correlation (r=0.94) between this method of measuring intrahepatic and total PSS and that evaluated by the more classic method using non-biodegradable microspheres.15
Intrarenal shunting was determined by a modification of the methodology used to determine PSS. Briefly, 20 μl (100 Mbq/ml) of 99mTc MDP was administered into the renal artery, the descending aorta distal to the renal artery being temporarily occluded by a snug ligature during injection of the isotopes. As 99mTc MDP is not retained within the kidney, the majority passes to the lungs and is registered graphically as the 100% passing fraction. The 99mTc MDP therefore acts as a reference injection. Following injection of 99mTc MDP, 20 μl (100 MBq/ml) of vortex mixed 99mTc albumin microspheres (mean diameter 20 μm) were administered into the renal artery. Unlike 99mTc MDP, the 99mTc albumin microspheres are trapped in the renal capillary bed and only those passing through the intrarenal arteriovenous shunts appear in the lung field of interest. The percentage of the passing fraction of99mTc albumin microspheres was expressed as a passing fraction of the reference injection of 99mTc MDP.
Hepatic, splanchnic, and renal blood flow was determined according to the method of McDevitt and Nies.16 In brief, the right femoral artery cannula was connected to a SAGE 351 (Orion Research Inc., Cambridge, Massachusetts, USA) withdrawal pump. A reference blood sample was withdrawn from the right femoral artery over a 90 second period at a rate of 0.6 ml/min. Ten seconds after the start of blood withdrawal each rat received an intraventricular injection of approximately 2–4 × 105cobalt-57 labelled carbonised microspheres (mean diameter 15 μm) suspended in 0.9% normal saline with 0.1% of Tween 80 (NEN-Trac, DuPont) in 0.6 ml over a period of 20 seconds using a constant rate infusion pump (SAGE 351, Orion Research Inc., Cambridge, Massachusetts, USA). Prior to injection the microspheres were sonicated for 10 minutes and then vortex mixed immediately before injection to ensure their complete disaggregation. To ensure that the withdrawal of a reference blood sample from the femoral artery cannula did not alter the arterial blood pressure, saline was infused through the left femoral venous cannula at the same rate as blood withdrawal.
After completion of all the studies the animals were killed with an anaesthetic overdose, and the organs were excised, weighed, placed in counting vials, and stored at 4°C for seven days until all the99mTc had decayed to an insignificant amount. The half life of 99mTc is six hours and consequently most will have decayed after 48 hours. In contrast, the half life of 57Co is 6520.8 hours and only a negligible decay of this isotope occurs before almost all the 99mTc has disappeared. The vials were placed in a Canberra Packard 500 automatic gamma well counter and the radioactivity counted. Cardiac output and organ blood flow were determined by the method of McDevitt and Nies.16
From the relative counts in the various organs the regional blood flow and cardiac output were calculated. The radioactivity of the reference blood sample was used to calculate the cardiac output (ml/min) using the formula:  Injected activity and reference activity are expressed as cpm and reference withdrawal rate as ml/min.
Injected activity and reference activity are expressed as cpm and reference withdrawal rate as ml/min.
Individual organ blood flow was determined using the formula: Organ activity and reference activity are expressed as cpm and organ blood flow and reference withdrawal rate as ml/min.
Organ activity and reference activity are expressed as cpm and organ blood flow and reference withdrawal rate as ml/min.
Portal venous inflow was calculated by adding together the flows to the stomach, small and large bowel, pancreas, mesentery, and spleen.
If the blood flow to the right and the left kidney differed by more than 10% the distribution of microspheres was not considered to be uniform and the results discarded.
BIOCHEMICAL STUDIES
Heparinised blood samples (0.2 ml) were obtained from the tail artery of all animals before administration of galactosamine (or saline) under light ether anaesthesia. Further blood samples were obtained from the femoral artery cannula 24, 36, 42, and 48 hours after galactosamine or saline administration. The samples were centrifuged at 4°C and 2000 g for 10 minutes and the plasma was analysed on a discrete multiple channel analyser (CXT Beckman, High Wycombe, UK).
AMINOPYRINE BREATH TEST
The aminopyrine breath test was used to assess liver function before and after intraperitoneal injection of galactosamine or saline. Each rat received 15 KBq of diethylamine-14C-aminopyrine (Amersham Corp., Arlington Heights, Illinois) in a volume of 100 μl intraperitoneally. The animals were housed in an airtight container through which air was pumped using a constant infusion pump. The exhaled14CO2 was collected in 15 minute breath samples for 120 minutes starting 60 minutes after aminopyrine administration.17 The radioactivity was counted in an Isocap 300 liquid scintillation system (Nuclear Data Inc., Medical Division, Chicago, Illinois, USA). The aminopyrine breath test constant (ABT-k) was calculated by least square regression analysis of the logarithm of counts versus time.18
STATISTICAL ANALYSIS
The results are presented as mean (SEM) together with 95% confidence intervals (CI). Comparisons between groups were made by one way analysis of variance (ANOVA) using a modified (Boneferroni)t test. Any correlation between changes in hepatosplanchnic haemodynamics and function and renal haemodynamics and function was determined using multiple regression analysis.
Results
LIVER FUNCTION TESTS
Plasma bilirubin and liver enzyme concentrations rose progressively in rats treated with galactosamine and were significantly increased (p<0.0001; ANOVA) above those observed in controls 24, 36, 42, and 48 hours after administration of the hepatotoxin (table 1). The increases in plasma concentrations of bilirubin and liver enzymes were maximal 42 hours after galactosamine administration and no further increases were observed thereafter (p=0.04; modifiedt test).
Liver function tests after injection of d-galactosamine or saline (control)
A progressive decrease (p<0.0001; ANOVA) in the ABT-k was observed 24, 36, 42, and 48 hours following galactosamine administration compared with controls (table 1). There was a highly significant correlation between the increases in the concentrations of serum bilirubin and liver enzymes and the reduction in the ABT-k (r=0.84, p<0.0001; multiple linear regression).
SYSTEMIC HAEMODYNAMICS
Although progressive liver failure induced by galactosamine was associated with decreases in the arterial blood pressure 36, 42, and 48 hours after administration of the hepatotoxin (table 2), the changes were not statistically significant (p=0.3; ANOVA). In contrast, there was a significant increase in cardiac output (fig 1) and a decrease in peripheral vascular resistance (table 2) 36, 42, and 48 hours following galactosamine administration compared with controls (p<0.001; ANOVA).
Circulatory and renal biochemical changes after injection of d-galactosamine or saline (control)
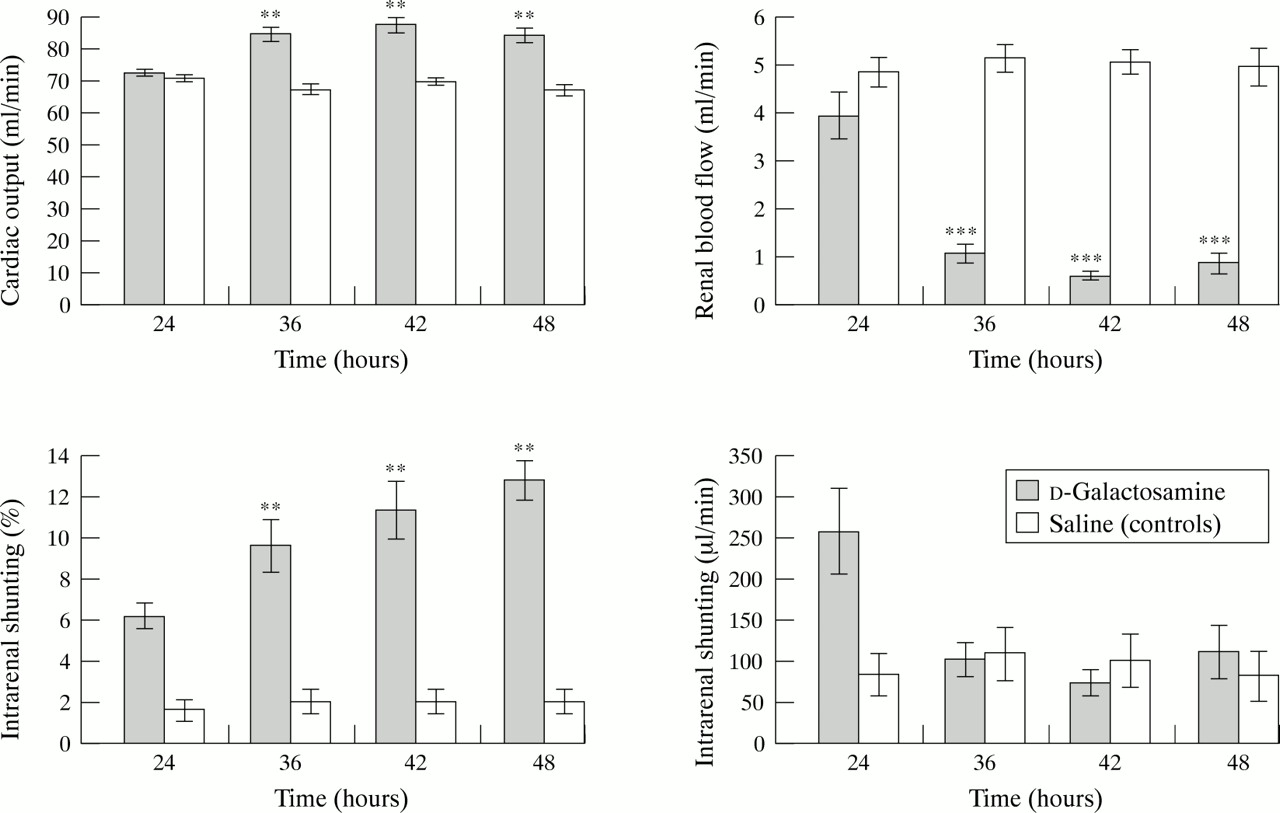
Changes in circulatory and renal haemodynamics after injection of d-galactosamine or saline. Results are expressed as mean (SEM). **p<0.01, ***p<0.001.
RENAL HAEMODYNAMICS AND FUNCTION
Liver injury was associated with a progressive impairment in renal function as evidenced by highly significant increases in serum urea and creatinine concentrations 36, 42, and 48 hours after galactosamine administration (table 2). Although cardiac output increased with progressive liver failure, there was a highly significant decrease in renal blood flow accompanied by a significant increase (p<0.0001; ANOVA) in intrarenal shunting (fig 1) 36, 42, and 48 hours after galactosamine administration. However, the absolute intrarenal shunt flow did not change significantly 36, 42, and 48 hours after galactosamine administration because of a significant decrease in renal blood flow (fig 1). The changes in renal haemodynamics were maximal 42 hours after galactosamine administration and no further significant changes (modified t test) were observed for the duration of the study.
HEPATOSPLANCHNIC HAEMODYNAMICS
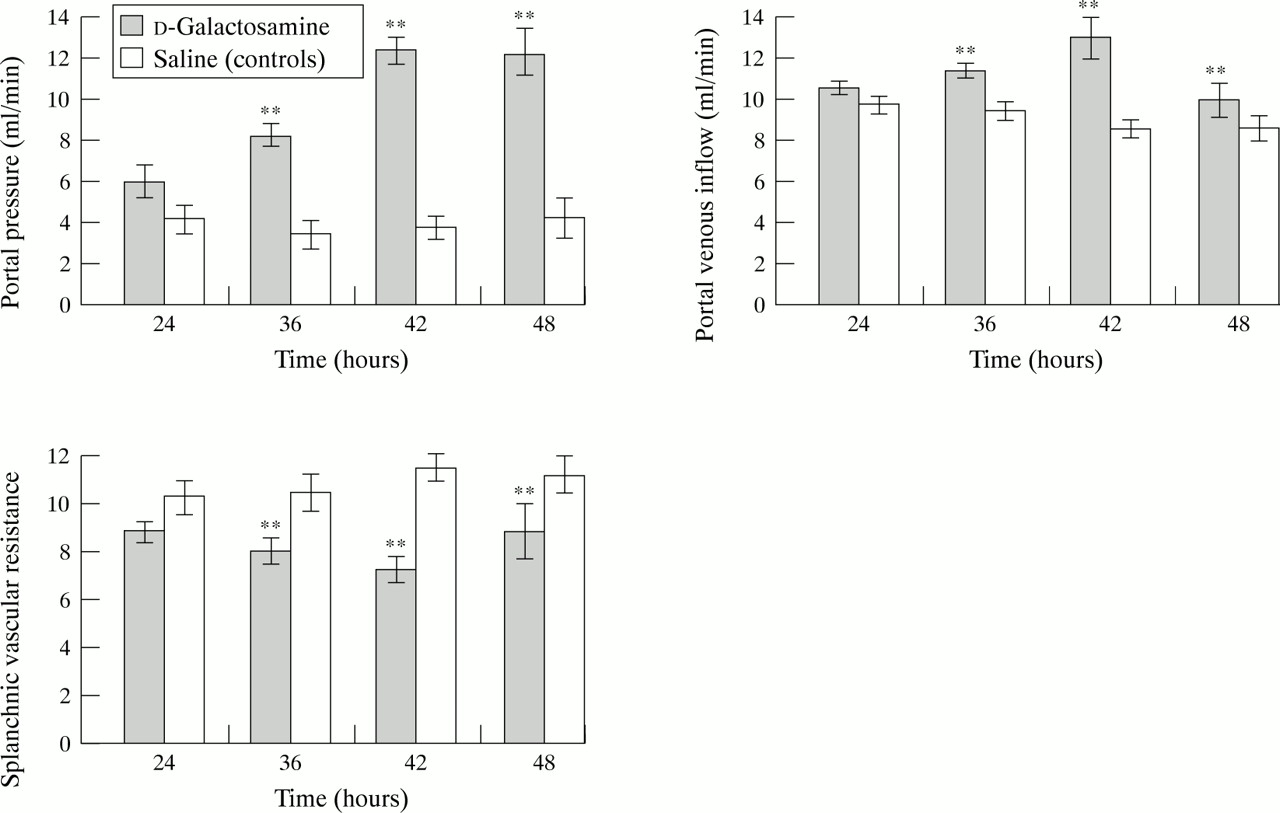
There were no significant changes in hepatic arterial flow in galactosamine treated rats compared with controls (table 3). In contrast, portal venous inflow and portal pressure were significantly increased and splanchnic vascular resistance decreased (p<0.001; ANOVA) 36, 42, and 48 hours following galactosamine administration compared with controls (fig 2). Furthermore, the degree of intrahepatic PSS was greatly increased (p<0.0001; ANOVA) 36, 42, and 48 hours following galactosamine administration compared with controls (fig 3). In contrast, there was no significant extrahepatic PSS in either control or galactosamine treated rats (table 3). The hepatosplanchnic haemodynamics were maximally altered 42 hours after galactosamine administration and no further significant changes occurred thereafter (modified t test).
Changes in hepatic arterial flow and extrahepatic portal-systemic shunting after injection of d-galactosamine or saline (control)

Changes in hepatosplanchnic haemodynamics after injection of d-galactosamine or saline. Results are expressed as mean (SEM). **p<0.01.

{kind=link}
{kind=link}
{kind=link}
Changes in intrahepatic portal-systemic shunting after injection of d-galactosamine or saline. Results are expressed as mean (SEM). ***p<0.001.
There was a highly significant correlation (r=0.84, p<0.0001; multiple linear regression) between the changes in renal blood flow and the increases in portal pressure (p=0.007) and intrahepatic PSS (p<0.0001). There was no significant correlation between the decrease in arterial blood pressure and the reduction in renal blood flow (p=0.3).
Discussion
Administration of the specific hepatotoxind-galactosamine to rats results in consistent toxic liver injury and liver failure.19 In the present study, progressive liver dysfunction was apparent 24 hours after administration of d-galactosamine as evidenced by increased plasma concentrations of liver enzymes and serum bilirubin and a decrease in plasma albumin concentrations. Maximal liver damage occurred 42 hours after administration of d-galactosamine and there were no further significant increases in plasma bilirubin concentrations and liver enzymes thereafter, for the duration of the study (48 hours). The aminopyrine breath test provides a quantitative measure of hepatic function in man and rats.20 ,21 In the present study, there was a positive correlation between changes in ABT-k and alterations in plasma concentrations of bilirubin and liver enzymes and a negative correlation with the decreased plasma albumin. The results of this study are therefore in accord with previous observations which suggest that d-galactosamine administration to rats results in the development of a robust and reproducible model of acute liver failure.14
Thirty six hours after administration of d-galactosamine, the rats displayed features of a hyperdynamic circulation with an increased cardiac output, arterial hypotension, and a decrease in systemic vascular resistance, observations in accord with previous studies.22 The development of hyperdynamic circulation in galactosamine treated rats was accompanied by a deterioration in renal function 36 hours after administration of galactosamine, as evidenced by increasing plasma concentrations of urea and creatinine. Furthermore, the alterations in renal function were accompanied by a highly significant decrease in renal blood flow 36 hours afterd-galactosamine administration, findings in accord with previous studies.23-25 The very large decrease in renal blood flow observed in the galactosamine treated rats could be due to either renal vasoconstriction6 or to a decrease in renal perfusion pressure.1 However, it seems unlikely that arterial hypotension was contributory to the observed decrease in renal perfusion as there was no significant correlation between changes in arterial blood pressure and renal haemodynamics. However, the reduction in renal perfusion in rats with galactosamine induced liver failure was accompanied by an increase in the relative degree of intrarenal shunting although the actual shunt flow did not change significantly. It has been suggested that renal hypoperfusion is accompanied by a redistribution of intrarenal blood flow.26 This hypothesis would appear to be supported by the results of the present study as renal hypoperfusion was accompanied by a significant increase in relative intrarenal shunting. However, the actual shunt flow did not change owing to renal hypoperfusion; only the percentage of total renal blood flow increased. These observations would suggest that during hypoperfusion, the size of the intrarenal shunts remains relatively constant; only the percentage of total blood passing through the shunt increases. Thus, the results of this study suggest that there is no redistribution of renal blood flow during hypoperfusion of the kidney, merely a relative increase in the percentage of blood passing through the intrarenal arteriovenous shunts.
The changes in renal haemodynamics observed in this study were associated with significant changes in hepatosplanchnic haemodynamics observed 36 hours after d-galactosamine administration. Thus, in this study galactosamine administration resulted in a significant increase in portal pressure and portal venous inflow, indicative of splanchnic pooling. This observation would support the hypothesis that in galactosamine induced liver failure, splanchnic pooling results in a decrease in effective blood volume, thereby reducing renal perfusion.27 There is however some controversy as to whether alterations in renal blood flow and function in liver disease are secondary to changes in hepatosplanchnic haemodynamics, in particular portal hypertension, or to deterioration in hepatic function. In experimental animals, a good correlation has been observed between the degree of portal hypertension and the reduction in renal perfusion which the authors attributed to a decreased glomerular filtration rate.28 In contrast, functional renal failure has never been correlated with the degree of portal hypertension in cirrhotic patients and furthermore, portacaval shunting does not improve renal function in the majority of patients undergoing such decompressive surgery.29 Other studies have shown that in cirrhotic rats, sodium retention, indicative of impairment of renal function, occurs when hepatic function decreases below a critical threshold.18 ,30 ,31 However, it should also be pointed out that in the aforementioned studies, the induction of cirrhosis also resulted in portal hypertension and changes in splanchnic haemodynamics which may also provoke sodium retention. In the present study, the increases in portal pressure were accompanied by changes in portal venous inflow and a deterioration in liver function; we were therefore unable to ascertain whether portal hypertension per se or alterations in liver function were primarily responsible for alterations in renal blood flow and function.
The most significant finding of this study was perhaps a highly significant increase in intrahepatic portal systemic shunting 36 hours after galactosamine administration, which as far as we are aware has not been previously described in acute liver failure and may play a major role in the alterations in renal blood flow and function. Thus, in cirrhosis and portal hypertension, gastrointestinal derived vasodilatory substances, such as glucagon, vasoactive intestinal polypeptide,32 substance P,33vasodilatory prostaglandins (PGE2 and prostacyclin),34 and endotoxins,35 which are normally removed by the liver, enter the systemic circulation via shunting through the collateral vessels. It is generally believed that an increase in the circulating levels of these gastrointestinally derived vasoactive agents in cirrhosis and portal hypertension may be responsible for peripheral vasodilatation and splanchnic pooling characteristic of this condition. These haemodynamic changes in cirrhosis would result in a decrease in the effective blood volume and therefore cause an increase in the sympathetic nervous system activity and activation of the renin-angiotensin axis, thereby inducing renal vasoconstriction.36 In addition, vasoconstrictor substances (leukotrienes and thromboxane B2) also enter the systemic circulation by a combination of PSS and hepatic dysfunction, further contributing to renal vasoconstriction.37 However, in acute liver failure, the duration of the disease has been usually considered to be too short for the development of a substantial collateral circulation. Therefore, it has been assumed that the gastrointestinal derived vasoactive agents responsible for the alterations in renal haemodynamics and function reach the circulation primarily as a result of impaired hepatocyte function.1 The results of this study clearly show that in acute liver failure, there is a large increase in intrahepatic PSS which may be a major mechanism whereby vasoactive agents reach the systemic circulation. This observation is supported by a significant correlation between intrahepatic PSS and the observed alterations in renal blood flow. Furthermore, the observation that intrahepatic PSS is markedly increased in acute liver failure has important potential therapeutic benefits. Thus administration of vasoactive drugs which markedly affect intrahepatic haemodynamics and which may decrease intrahepatic PSS may ameliorate not only perturbations in renal blood flow and function but also other abnormalities characteristic of this syndrome such as hepatic encephalopathy and systemic circulatory abnormalities.
In summary, the results of this study suggest that increased intrahepatic portal-systemic shunting and hepatocyte impairment may initiate a chain of events causing alterations in the systemic levels of vasoactive agents which culminate in alterations in renal haemodynamics and function.
Acknowledgments
This work was supported by a grant from the Mersey Kidney Research Fund. The work was presented at the Annual Meeting of the Surgical Research Society, Manchester, July 1995, and published in abstract form in the British Journal of Surgery (1995;82:1565–6).