Article Text

Abstract
Background—Basic fibroblast growth factor (bFGF) promotes angiogenesis and healing of gastric ulcers in rats, and bFGF expression is up regulated in such ulcers. However, little is known about expression of bFGF in human gastric mucosa.
Aims—To investigate bFGF expression in intact human gastric mucosa and gastric ulcers and to determine whether low bFGF content or altered binding by mucosa is associated with ulceration.
Subjects—Endoscopy outpatients, gastrectomy patients, and organ donors.
Methods—bFGF was isolated by heparin affinity chromatography and characterised by western blotting and endothelial cell bioassay. bFGF was measured by immunoassay and its distribution defined by immunohistochemistry and in situ hybridisation. Binding of bFGF by heparan sulphate proteoglycans was investigated by sodium chloride and heparin extraction.
Results—Bioactive bFGF (19 kDa) was detected in normal mucosa but bFGF mRNA was not found. bFGF expression was up regulated in granulation tissue endothelial cells, mononuclear cells, and epithelial cells at the ulcer rim. Gastric ulcer patients had constitutively low bFGF concentrations in intact antral mucosa which were not explained by changes in binding to heparan sulphate proteoglycans.
Conclusions—bFGF expression is up regulated in human gastric ulcers. Low intact mucosal bFGF content is associated with gastric ulceration.
- basic fibroblast growth factor
- gastric mucosa
- heparan sulphate proteoglycan
- peptic ulceration
Statistics from Altmetric.com
Basic fibroblast growth factor (bFGF) is a protein which stimulates angiogenesis and accelerates healing of experimental gastric and duodenal ulcers in rats.1 ,2 bFGF has been given to patients with non-steroidal anti-inflammatory drug (NSAID) associated gastric ulcers in an open, pilot study. At four weeks, there was a 44% healing rate and a mean 89% decrease in area of unhealed gastric ulcers.3
Despite evidence that exogenous bFGF promotes healing of gastric ulcers, there is little evidence for a role for endogenous mucosal bFGF in gastric ulcer repair. Increased expression of bFGF has been noted in granulation tissue of gastric1 and duodenal ulcers4 in rats and a neutralising anti-bFGF antibody delayed healing of gastric ulcers in the same rat model.1bFGF has been isolated from human gastric mucosa, obtained after gastric carcinoma resection,4 and two groups have reported the immunohistochemical localisation of bFGF in normal gastric mucosa with conflicting results5 ,6; there are however no data on bFGF expression in gastric ulcer patients. bFGF is believed to be bound by heparan sulphate proteoglycans (HSPGs) which may modulate bFGF activity at the cell surface,7 protect bFGF from inactivation by acid and proteolysis,8 and act as a “storage site” of preformed bFGF for release following tissue injury.9
In this study, we investigated bFGF expression and binding by mucosal HSPGs in intact and ulcerated human gastric mucosa. We tested the hypothesis that decreased mucosal bFGF content or altered binding to HSPGs is associated with ulceration and that bFGF expression is up regulated in human gastric ulcers.
Materials and methods
ETHICAL APPROVAL
Written permission to collect endoscopic gastric biopsy specimens from outpatients undergoing upper gastrointestinal endoscopy, gastric ulcer resection specimens from patients undergoing laparotomy for haemorrhage or perforation, and whole stomachs from organ donors was obtained from patients and relatives. Studies were approved by the University Hospital Ethics Committee.
ISOLATION AND CHARACTERISATION OF GASTRIC MUCOSAL bFGF
Heparin affinity chromatography
Gastric mucosa was obtained from organ donor stomachs by dissection and weighed. Mucosa was homogenised on ice for two minutes in 4 ml extraction buffer (10 mmol/l Tris-Cl (pH 7.4), 2 mol/l NaCl, 1 mmol/l EDTA, 1 mmol/l phenylmethylsulphonyl fluoride, 1 μg/ml aprotinin, and 1 μg/ml pepstatin A (all Sigma, Poole, UK)) per gram of mucosa. The homogenate was sonicated for 30 seconds × 5 and stirred overnight at 4°C. The homogenate was centrifuged (2000g, 15 minutes at 4°C) to remove tissue debris and then ultracentrifuged (15 000 g, 30 minutes at 4°C). Decanted supernatant was diluted ×4 with 10 mmol/l Tris-Cl (pH 7.4).
A 5 ml heparin-sepharose column (Pharmacia Biotech Ltd, St Albans, UK), equilibrated with 25 ml 10 mmol/l Tris-Cl (pH 7.4) was connected to a low pressure chromatography system (Econo system, Bio-Rad Laboratories Ltd, Hemel Hempstead, UK) and the supernatant added. The column was washed with 10 mmol/l Tris-Cl (pH 7.4), 0.6 mol/l NaCl until the eluate read background absorbance at 280 nm. Heparin bound proteins were eluted using an 80 ml gradient of 0–3 mol/l NaCl in 10 mmol/l Tris-Cl (pH 7.4). Forty 2 ml fractions were collected and equilibrated with 10 mmol/l Tris-Cl (pH 7.4), 0.15 mol/l NaCl using 9 ml Sephadex G-25 columns (Pharmacia) before assay for bFGF.
bFGF enzyme linked immunosorbent assay (ELISA)
The bFGF ELISA was similar to that previously described by Watanabe et al.10 Briefly, samples or standards (100 μl) were added to an equal volume of 100 μg/ml porcine heparin (Sigma) in blocking solution (25% Block Ace (Wako Chemicals, Osaka, Japan) in phosphate buffered saline (PBS)) and 100 μl aliquots were added in duplicate to wells coated with two murine monoclonal antirecombinant human bFGF antibodies (Mab52 and Mab98; Wako) at 5 μg/ml. After overnight incubation at 4°C and washing with deionised water, the Fab′ fragment of a third murine monoclonal anti-bFGF antibody (Mab3H3; Wako) conjugated to horseradish peroxidase (HRP) in blocking solution was added to wells at 3 μg/ml and incubated for two to four hours at room temperature. Following further washes with deionised water and PBS, o-phenylenediamine dihydrochloride (2 mg/ml, Sigma) and 0.03% H2O2 were added to each well and incubated in the dark at room temperature for 60 minutes. The reaction was terminated by addition of 2 mol/l H2SO4. Absorbance at 492 nm was measured using a Dynatech MR5000 microplate reader.
There was no cross reactivity with FGF1, 4, 5, 6, or keratinocyte growth factor (FGF7) using this ELISA (own data, not shown; see also Watanabe et al 10).
Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE)
Aliquots (500 μl) from heparin affinity chromatography fractions were pooled, freeze dried, and reconstituted in 100 μl double distilled (dd) H2O. An equal volume of 2× electrophoresis sample buffer (0.125 mol/l Tris-Cl (pH 6.8), 4% SDS, 10% mercaptoethanol, 20% sucrose, 0.004% bromophenol blue; all Sigma) was added to 7.5 μl of pooled eluate and boiled for five minutes before cooling on ice. Samples were electrophoresed on 12.5% 29/1 acrylamide/bis-acrylamide minigels along with molecular weight markers (SDS-PAGE Broad Range Molecular Weight Standards, Bio-Rad) and 7.5 μl 250 μg/ml carrier free recombinant bFGF corresponding to amino acid residues 9–155 of human bFGF (R&D Systems Europe, Abingdon, UK). Minigels were fixed in 20% methanol/7.5% acetic acid and stained with 0.1% Coomassie brilliant blue R-250 (Bio-Rad) as well as a silver stain (Silver Stain Plus, Bio-Rad).
Western blot analysis
Following SDS-PAGE, proteins were blotted onto 0.2 μm nitrocellulose membrane (Bio-Rad) using a semidry transfer cell (Bio-Rad). The membrane was blocked with 3% gelatin in Tris-buffered saline (TBS) for 60 minutes and washed with 0.05% Tween-20 (Sigma) in TBS before incubation with 1 μg/ml goat polyclonal antihuman bFGF antibody (R&D Systems) at 4°C overnight. After further washes with 0.05% Tween-20, the membrane was incubated with a 1/2000 dilution of rabbit antigoat IgG-HRP (Sigma) at room temperature for 60 minutes before addition of substrate (4-chloro-1-napthol/H2O2; Bio-Rad).
Bioassay of gastric mucosal bFGF
Heparin affinity chromatography fractions were pooled, freeze dried, and resuspended in 500 μl dd H2O. This solution was exchanged for 10 mmol/l Tris-Cl (pH 7.4), 0.15 mol/l NaCl using gel filtration (Bio-Spin 6, Bio-Rad) and sterilised using a 0.2 μm filter (Sartorius AG, Gottingen, Germany).
A 95 μl aliquot of this solution or 10 mmol/l Tris-Cl (pH 7.4), 0.15 mol/l NaCl were preincubated with 5 μl of 1 mg/ml Mab3H3 or 5 μl PBS for 60 minutes at room temperature. Mab3H3 has previously been shown to neutralise the effects of bFGF in vivo and in vitro11 and we have shown that the concentration used here (50 μg/ml) inhibits the proliferative activity of 10 ng/ml recombinant human bFGF on human umbilical vein endothelial cells (HUVECs; data not shown). The mitogenic activity of these samples on human gastric endothelial (HuGE) cells was measured using a colorimetric proliferation assay as previously described.12
IMMUNOHISTOCHEMISTRY FOR bFGF
Formalin fixed, paraffin wax embedded sections (4 μm thick) of gastric mucosa and Dupytren’s contracture (used as a positive control)13 were dewaxed and rehydrated. Endogenous peroxidase activity was blocked with 4 parts methanol/1 part 3% H2O2 for 20 minutes. After washing in PBS, sections were blocked with 10% rabbit serum in PBS for 60 minutes at room temperature. Sections were incubated with Mab52 (6 μg/ml) diluted in 10% rabbit serum at 4°C overnight. Mab52 did not cross react with FGF1, 4, 5, 6, or 7 up to 1 ng/ml when tested by direct ELISA (data not shown).
Following PBS washes (3 × five minutes), sections were incubated with a 1/400 dilution of biotinylated polyclonal rabbit antimouse immunoglobulin antibody (Dako Ltd, High Wycombe, UK) and visualised using streptavidin-biotin-HRP (Dako) and 3,3′-diaminobenzidine/H2O2 (both 0.7 mg/ml; Sigma). Controls comprised omission of the primary antibody, substitution of the primary antibody with a non-specific mouse IgG2b monoclonal antibody (anti-Aspergillus niger glucose oxidase; Dako) and preabsorption of the primary antibody with ninefold molar excess of bFGF for two hours at 4°C before use.
Mucosal bFGF content was assessed using a semiquantitative scoring system by an experienced histopathologist (DJ) blind to the origin of the sections and the corresponding bFGF concentration. Epithelial cell, lamina propria, smooth muscle, and muscularis mucosae staining was scored on a scale from 0–3 (0, absent staining; 1, weak staining, just visible; 2, moderate staining, easily seen; 3, strong staining). A total bFGF score was obtained by addition of individual components. Adjacent sections stained with haematoxylin and eosin (H&E) were assessed independently for acute and chronic inflammatory infiltration, atrophy, and reactive gastritis (foveolar hyperplasia, presence of dilated, superficial capillaries, and lamina propria smooth muscle infiltration) using a scale of 0–3 by the same histopathologist.
IN SITU HYBRIDISATION OF bFGF mRNA
Preparation of bFGF cDNA probe
Plasmid pTB669 was a kind gift from Dr Toguchi (Pharmaceutical Research Division, Takeda). Plasmid pTB669 contains a 450 bp cDNA insert corresponding to the coding sequence for amino acid residues 4–146 of human bFGF.14
bFGF cDNA was excised using EcoRI and BglII (Northumbria Biologicals Ltd, Cramlington, UK). bFGF cDNA and pBluescript II SK DNA (SK II, Stratagene, La Jolla, California, USA) were labelled with35S-dCTP (1000–1500 Ci/mmol, Amersham, Little Chalfont, UK) or 32P-dCTP (3000 Ci/mmol, Amersham) for in situ hybridisation or northern blot analysis respectively using a random priming technique (Nonaprimer II, Appligene, Birtley, UK). Labelled probes were purified using silica gel chromatography (QIAGEN Ltd, Dorking, UK).
Northern blot analysis
Gastric mucosa from the antrum of two organ donor stomachs was snap frozen at −70°C immediately after excision. Total RNA was extracted by a guanidium isothiocyanate-phenol-chloroform extraction technique (Trizol, GIBCO BRL, Paisley, Scotland, UK) based on the method of Chomczynski and Sacchi.15 Poly A+RNA was obtained using poly dT affinity chromatography (QIAGEN). Poly A+ RNA (1 μg) was electrophoresed and blotted onto Hybond-N nylon membrane (Amersham) by capillary transfer overnight and then crosslinked to the membrane using ultraviolet light. The membrane was hybridised with 106 cpm/ml 32P-dCTP labelled bFGF cDNA probe in 5× SSPE, 5× Denhardt’s solution (Sigma), 0.5% SDS at 65°C overnight. The final stringency wash was 2 × 20 minutes in 0.1× SSPE, 0.1% SDS at 50°C. Autoradiography was for 96 hours at −70°C using Kodak X-Omat AR film (Sigma) and two intensifying screens.
Confirmation that the bFGF cDNA probe hybridised with known bFGF mRNA species was obtained by northern blot analysis of 1 μg poly A+ from human omental microvascular endothelial (HOME) cells and HUVECs.
In situ hybridisation
The method used was similar to that of Sandberg and Vuorio.16 Sections (4 μm) of formalin fixed, paraffin wax processed gastric mucosa and Dupytren’s contracture were dewaxed and rehydrated. Sections were incubated in 0.2 N HCl for 20 minutes and then washed in 2× SSC (standard saline citrate; 1× SSC = 0.15 mol/l NaCl, 0.015 mol/l trisodium citrate) at 70°C for 10 minutes before digestion with 10 μg/ml proteinase K (Sigma) for 15 minutes at 37°C. Sections were then acetylated with 0.25% acetic anhydride in 0.1 mol/l triethanolamine (pH 8) for 10 minutes. Sections were hybridised overnight at 42°C with 105–106cpm/section of 35S labelled bFGF cDNA probe in 50% formamide, 10 mmol/l dithiothreitol (DTT), 1× Denhardt’s solution, 0.6 mol/l NaCl, 10 mmol/l Tris, 2 mmol/l EDTA, 10% dextran sulphate, and 3% salmon sperm DNA (Sigma). Negative control sections were incubated with 35S labelled SK II DNA of similar activity. Sections were given stringency washes in 0.5× SSC, 1 mmol/l EDTA, 10 mmol/l DTT at 42°C for 10 minutes followed by 2 × 10 minute washes with 0.5× SSC, 1 mmol/l EDTA at 42°C. Sections were then washed with 50% formamide, 0.15 mol/l NaCl, 0.5 mmol/l EDTA, 5 mmol/l Tris-Cl (pH 7.4) at room temperature for 10 minutes. Four five minute washes with 0.5× SSC at 55°C were followed by a wash with 0.5× SSC at room temperature. The probe was localised using Hypercoat LM-1 emulsion (Amersham) autoradiography with exposure for up to 28 days at 4°C. Sections were counterstained with haematoxylin.
MEASUREMENT OF GASTRIC MUCOSAL bFGF CONCENTRATIONS
Upper gastrointestinal endoscopy using an Olympus PQ-20 or XP-10 endoscope was performed on outpatients who attended routine endoscopy lists or an NSAID screening programme. A gastric ulcer was defined as having a diameter of greater than 3 mm and perceptible depth. Four biopsy specimens were taken from the ulcer rim if present and four from macroscopically normal antral mucosa at least 2 cm distant from the ulcer using forceps with a 5 mm bite. Ulcer rim biopsy specimens were taken at 45° from the mucosal surface in an attempt to include ulcer base and intact mucosa in equal proportions. Two further biopsy specimens from intact antral mucosa were placed in formalin, stained with H&E, and examined for acute and chronic inflammatory infiltration, atrophy, and reactive gastritis as described. A final antral biopsy specimen was taken for a rapid urease test. Age, sex, smoking status, past history of gastric ulceration, ulcer healing drug use, and NSAID use were noted at the same time. An NSAID user was defined as one taking NSAIDs regularly more than three days per week.
Biopsy specimens were immediately placed in 1 ml 10 mmol/l Tris-Cl (pH 7.4), 0.15 mol/l NaCl, vortexed briefly, and pulse centrifuged. The biopsy specimens were removed from the wash solution and dried briefly on filter paper before weighing. Specimens were placed in 600 μl fresh 10 mmol/l Tris-Cl (pH 7.4), 0.15 mol/l NaCl and homogenised for 60 seconds using an Ultra-Turrax T25 homogeniser. Homogenates were immediately centrifuged at 15 000 g for 60 seconds. The supernatant was collected and added to an equal volume of PBS containing 100 μg/ml heparin and 25% Block Ace. Aliquots (100 μl) of this solution were added to a bFGF ELISA plate. Results are expressed as pg bFGF per mg wet weight of biopsy specimen. Intra-assay coefficient of variation of the ELISA was 6.1% (mean bFGF concentration 94.0 pg/well; n=8). Interassay coefficient of variation was 22.0% (mean bFGF concentration 19.0 pg/well; n=4). Recovery of added recombinant bFGF (2 pg/well) to homogenate supernatant was 130%.
bFGF EXTRACTION FROM GASTRIC MUCOSA BY NaCl AND HEPARIN
Two sets of four biopsy specimens were obtained from other patients undergoing upper gastrointestinal endoscopy and were washed, weighed, and homogenised in 600 μl 10 mmol/l Tris-Cl (pH 7.4), 0.15 mol/l NaCl as described above except that homogenates were incubated for 10 minutes on ice before centrifugation. Homogenates were centrifuged at 15 000 g for 60 seconds and supernatants were collected. One of the pair of gastric mucosal pellets was resuspended in 600 μl ice cold 10 mmol/l Tris-Cl (pH 7.4), 0.5 mol/l NaCl, vortexed for 60 seconds, and left on ice for 10 minutes. The sample was centrifuged and the supernatant collected. This process was repeated with separate 600 μl aliquots of 10 mmol/l Tris-Cl (pH 7.4) with added 1.0 mol/l, 1.5 mol/l, and 2.0 mol/l NaCl. Remaining bFGF in the pellet was extracted using 600 μl of 100 μg/ml heparin (Sigma) in 10 mmol/l Tris-Cl (pH 7.4), 0.15 mol/l NaCl. Finally, extraction of residual bFGF was performed using 10 mmol/l Tris-Cl (pH 7.4), 3.0 mol/l NaCl.
All supernatants were buffer exchanged with 10 mmol/l Tris-Cl (pH 7.4), 0.15 mol/l NaCl using Biospin 6 columns (Bio-Rad). Samples were stored at −30°C until assay for bFGF by ELISA.
At the same time, the other mucosal pellet (control) was handled in a similar fashion except that increasing NaCl concentrations were replaced by repeated 600 μl aliquots of 10 mmol/l Tris-Cl (pH 7.4), 0.15 mol/l NaCl. After this, incubation with 100 μg/ml heparin solution followed by 10 mmol/l Tris-Cl (pH 7.4), 3.0 mol/l NaCl was carried out as above. Buffer exchange was also performed on control supernatants. Salt and control extracts were assayed in duplicate by bFGF ELISA.
STATISTICAL ANALYSIS
Gastric mucosal homogenate bFGF concentrations did not have a normal distribution and so were logarithmically transformed before statistical analysis by Student’s t test for paired and unpaired data. Data are expressed as the geometric mean and 95% confidence interval (CI). bFGF histology and gastritis scores were compared using the Mann-Whitney U test. Multivariate analysis of variance was used to determine whether Helicobacter pyloristatus, NSAID use, or type and degree of gastritis accounted for differences in bFGF concentrations and bFGF histology scores between patient groups.
bFGF concentrations extracted from gastric mucosa using NaCl and heparin are expressed as the arithmetic mean and standard error of the mean (SEM) and differences were analysed using Student’st test.
Results
bFGF PROTEIN IN NORMAL HUMAN GASTRIC MUCOSA
Isolation and characterisation of gastric mucosal bFGF
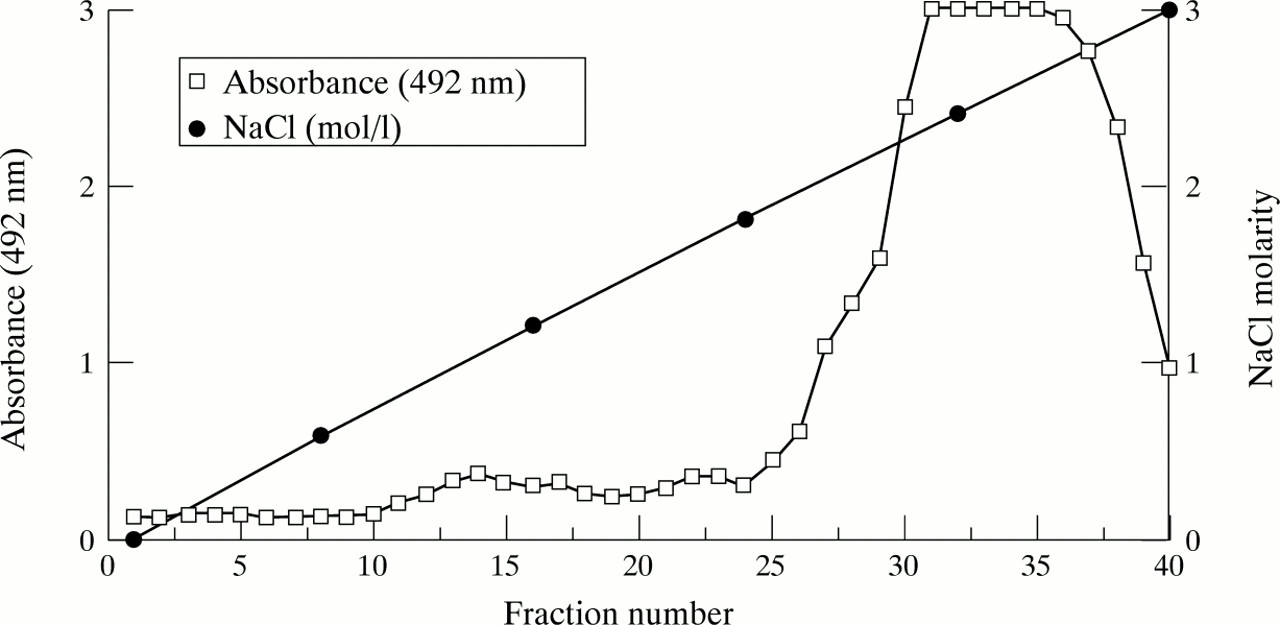
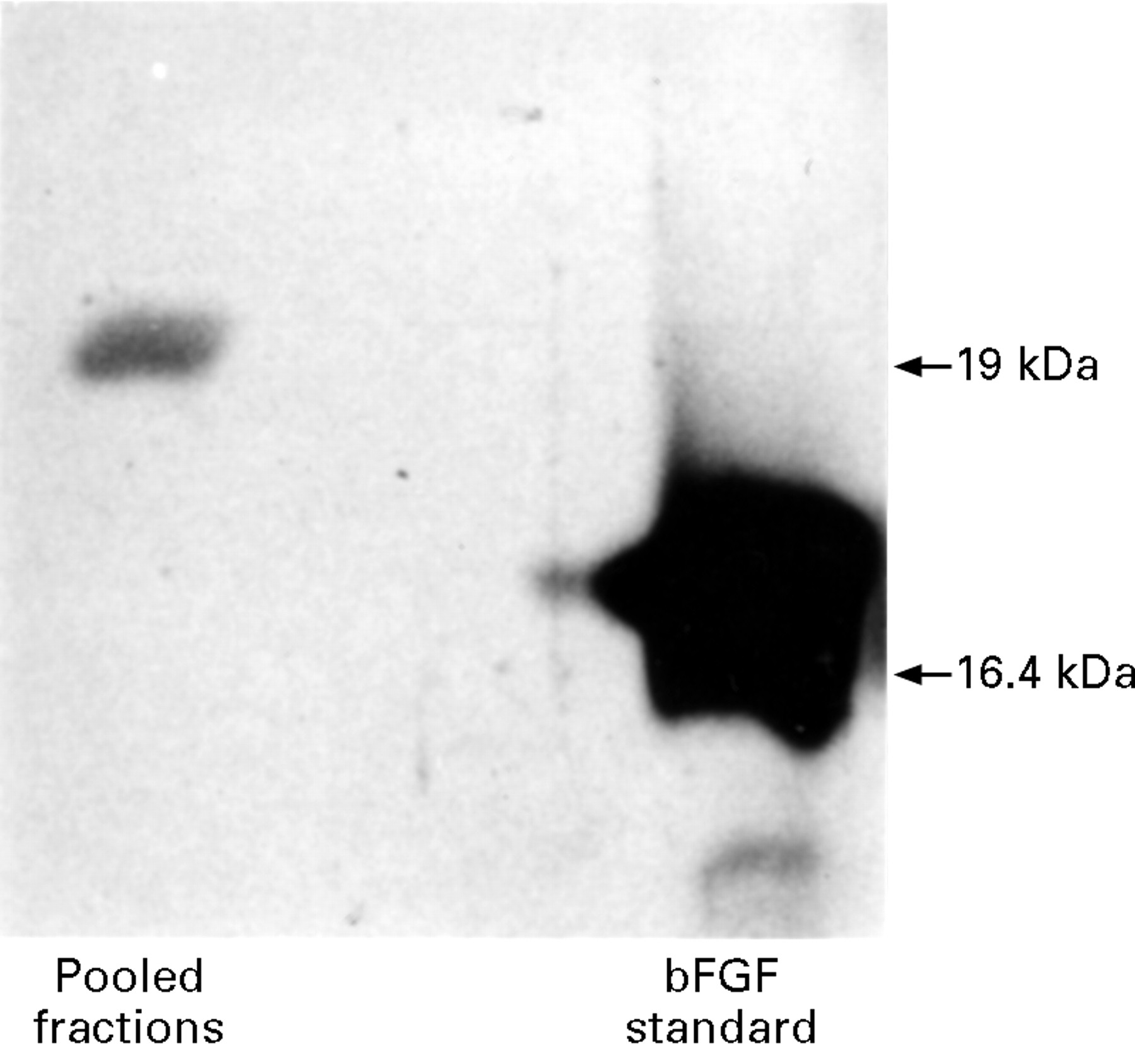
Basic FGF eluted from heparin-sepharose between 1.8 mol/l and 2.8 mol/l NaCl (fig 1). The elution profile was similar from three organ donor stomachs. Western blot analysis of pooled fractions 30–40 revealed a single 19 kDa bFGF isoform (fig 2). Silver staining of the pooled fractions showed no contaminating protein. Gastric mucosal bFGF, isolated in this manner, stimulated proliferation of HuGE cells (fig3). Preincubation with Mab3H3 inhibited the proliferative response to gastric mucosal bFGF by more than 80% (fig 3).

Elution of bFGF from human gastric mucosa using heparin affinity chromatography and a 0–3 mol/l NaCl gradient. bFGF content of each fraction was assessed using a bFGF ELISA. A peak of bFGF was detected in fractions 30–40 corresponding to elution by 1.8–2.8 mol/l NaCl.

Western blot analysis of gastr ic mucosal bFGF. Fractions 30–40 from heparin affinity chromatography were pooled, freeze dried, and reconstituted in 100 μl H2O. Gastric mucosa contained a 19 kDa isoform of bFGF compared with 16.4 kDa N-terminal truncated recombinant bFGF.

Mitogenic activity of gastric mucosal bFGF. HuGE cell number was assayed using a formazan colorimetric assay (Promega, Madison, Wisconsin). We have previously shown a good correlation (r2=0.99) between absorbance at 492 nm and HuGE cell number using this assay.12 Briefly, 103 HuGE cells were added to individual 1% gelatin coated wells of a 96 well plate and incubated for 48 hours in 5% CO2 at 37°C in the absence of endothelial cell growth supplement. Gastric mucosal bFGF or control buffer with or without Mab3H3 (working concentration 50 μg/ml) was added to HuGE cell monolayers and incubated for a further 24 hours before assay. Data are expressed as the mean (SEM) multiple of the absorbance at 492 nm of the negative control (n=4 wells). Gastric mucosal bFGF promoted proliferation of HuGE cells which was almost completely inhibited by Mab3H3. Cell number was not affected by the presence of Mab3H3 per se (data not shown).
Immunohistochemistry
Sections of normal gastric antral and body mucosa from organ donors, who were not taking NSAIDs, were used. There was no histological evidence of stress ulceration or H pyloriinfection in these cases.
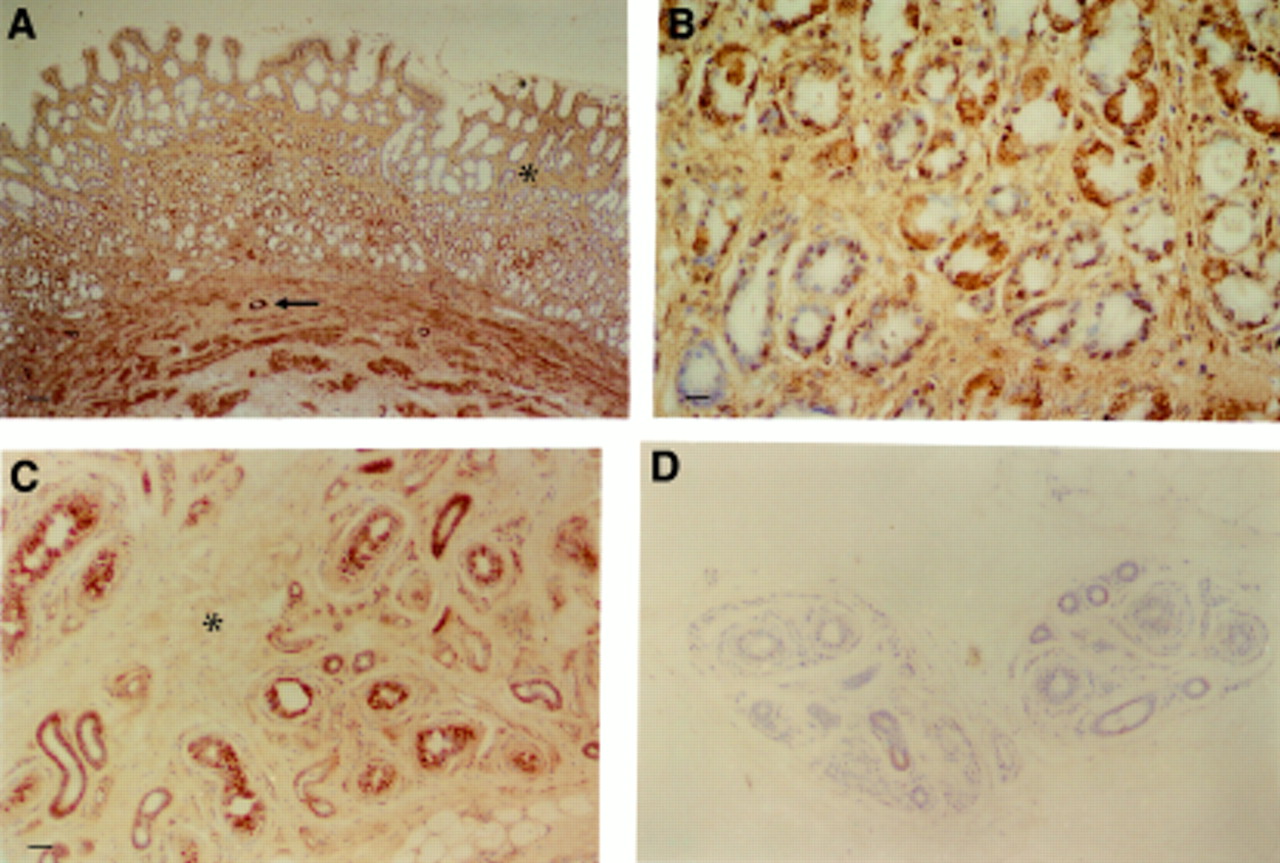
In antral mucosa, staining of bFGF was present in glandular epithelial cells and surface epithelium (fig 4A). In gastric glands, the distribution of bFGF was patchy. Staining appeared to be diffusely cytoplasmic (fig 4B) although a few cells had some punctate nuclear staining. Basic FGF immunoreactivity was also detected in extracellular matrix surrounding lamina propria cells and glands (fig 4A). There was also staining of muscularis mucosae and endothelium of arterioles (fig4A). Endothelial cells of microvessels in the lamina propria contained less immunoreactive bFGF than those of larger vessels.

Immunohistochemistry for bFGF in normal tissue. (A) Normal antral mucosa. Localisation of bFGF to surface and glandular epithelium, lamina propria extracellular matrix (ECM) (asterisk), muscularis, and blood vessels (arrow). Bar=80 μm. (B) Normal antral mucosa. Patchy distribution of bFGF in glandular epithelium. Bar=20 μm. (C) Dupytren’s contracture. Staining of sweat glands, blood vessels, and ECM (asterisk). Bar=40 μm. (D) Dupytren’s contracture. Preabsorption of Mab52 by recombinant human bFGF (×9 molar excess). No staining. Scale as for (C).
In body mucosa, there was a similar pattern of staining but glandular epithelium was less intensely stained than antral epithelium.
There was specific staining of sweat glands, blood vessels, and occasional myofibroblasts in Dupytren’s contracture (fig 4C) in agreement with published data on the distribution of bFGF in this tissue.13 ,17 Control sections of Dupytren’s contracture and gastric mucosa revealed no staining (fig 4D).
bFGF mRNA IN NORMAL HUMAN GASTRIC MUCOSA
bFGF mRNA was not detected in normal human gastric mucosa by northern blot analysis although the bFGF probe detected three bFGF mRNAs in HOME cells (6.6, 2.0, and 1.0 kb) and one in HUVECs (6.6 kb; data not shown) confirming previous data.18 Similarly, there was no hybridisation of the bFGF probe with sections of normal antral and body mucosa although specific signal was detected over occasional fibroblastoid cells and arteriolar smooth muscle of Dupytren’s contracture as described previously.13 ,17 No specific hybridisation was shown with the control (SK II) probe.
LOW bFGF CONCENTRATIONS IN INTACT GASTRIC MUCOSA OF ULCER PATIENTS
Next, we investigated whether differences in bFGF expression in intact mucosa were associated with the presence of gastric ulceration. Intact mucosal bFGF concentrations from 17 gastric ulcer patients were compared with corresponding values from 52 age and sex matched non-ulcer control patients. Table 1 shows that these two groups were well matched for NSAID use, H pylori, and smoking status. bFGF concentrations in intact antral mucosa of ulcer patients (mean 10.4 pg/mg; 95% confidence interval (CI) 5.3–15.5) were notably lower than in intact mucosa of non-ulcer controls (234.0 pg/mg; CI 174.0–294.0) (p< 0.001; fig 5). Lower intact mucosal bFGF concentrations in gastric ulcer patients were not explained by differences in NSAID use, H pylori status, or type and degree of gastritis between ulcer patients and non-ulcer controls when assessed by multivariate analysis of variance (table2).
Gastric ulcer patients and non-ulcer controls from whom endoscopic biopsy samples were taken

Intact mucosal bFGF concentrations in gastric ulcer patients and matched non-ulcer controls. *p<0.001.
Multivariate analysis of variance of factors which could account for the low intact gastric mucosal bFGF concentrations measured by ELISA
Repeat antral biopsy specimens were obtained from the antrum of eight gastric ulcer patients at eight weeks, at which time complete ulcer healing had occurred in all cases. Acid suppressing drugs were still being used at the time of endoscopy (three patients received standard healing doses of an H2 receptor antagonist and five patients were enrolled into a blinded study of omeprazole 20 mg or 40 mg daily or misoprostol 200 μg four times daily for healing of NSAID associated gastric ulcers). Intact mucosal bFGF concentrations rose significantly following ulcer healing with acid suppressing drugs (mean 8.9 (CI 1.8–16.0) pg/mg prehealing versus 41.5 (CI 12.3–70.7) pg/mg posthealing; p=0.02) but concentrations did not reach those seen in non-ulcer control patients (fig 5).
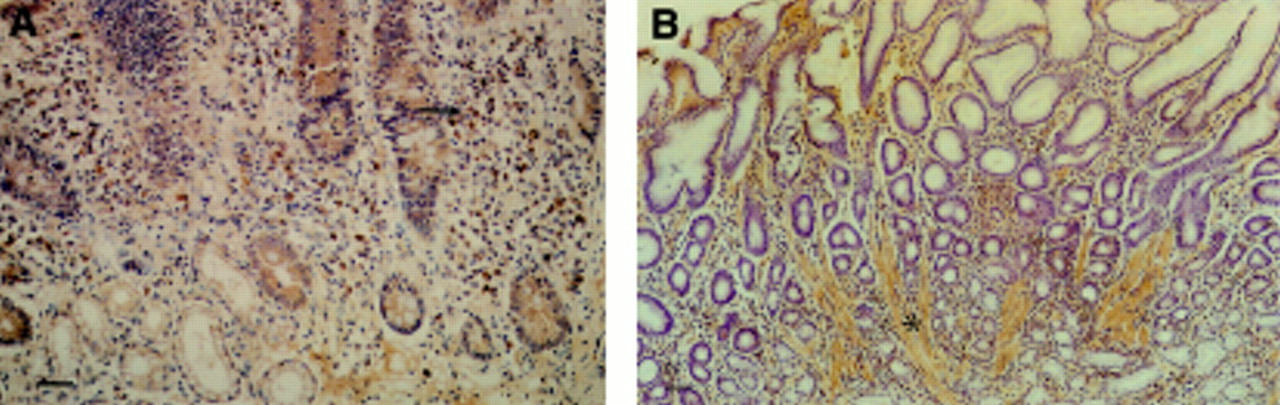
Histological assessment of bFGF content in intact antral mucosa was carried out in 10 gastric ulcer patients and eight non-ulcer controls which were matched for age, sex, H pylori status, and NSAID use. The distribution of bFGF was similar to that described in normal gastric mucosa. In cases in which a chronic inflammatory infiltrate was present, an increase in lamina propria mononuclear cells positive for bFGF was seen (fig 6A). Superficial lamina propria muscle fibres in gastric mucosa showing other features of reactive gastritis stained positively for bFGF (fig 6B). There was a strong positive correlation between the degree of reactive gastritis and the bFGF histology score in individual patients (r=0.75, p<0.001; Spearman rank correlation). Otherwise, there was no effect of the type and degree of gastritis on bFGF immunostaining. Antral mucosa from gastric ulcer patients had a significantly higher bFGF histology score (and degree of reactive gastritis) than mucosa from non-ulcer patients despite having notably lower bFGF concentrations measured by ELISA (table 3).

Immunohistochemistry for bFGF in antral gastric mucosa. (A) H pylori associated gastritis. Strongly stained mononuclear cells present in a chronic inflammatory infiltrate (arrow). Bar=50 μm. (B) Reactive (type C) gastritis. Prominent lamina propria smooth muscle fibres positive for bFGF (asterisk). Bar=100 μm.
Comparison of bFGF concentrations and bFGF histology score in patients with gastric ulcer and non-ulcer controls
BINDING OF bFGF TO GASTRIC MUCOSAL HSPGs
We then investigated whether alterations in strength of binding of bFGF to gastric mucosal HSPGs could explain low bFGF concentrations measured by ELISA in gastric ulcer patients in the absence of reduced immunohistochemical staining. Graded NaCl and heparin extraction of bFGF was performed on mucosa from 15 different patients with an active ulcer (n=7) or a healed ulcer (n=8) and 12 new control (non-ulcer) patients (table 4). The two groups were well matched except for age (non-ulcer patients were younger) and for use of acid suppressing drugs (greater in ulcer patients). Four patients in each group were taking a proton pump inhibitor which did not allow accurate assessment ofH pylori status.
Patients with gastric ulcer and controls who underwent biopsy of the antrum for bFGF extraction
bFGF was extracted from antral mucosa in two peaks (fig 7A); 30% of total bFGF eluted at 0.15 mol/l NaCl (peak 1) and 23% of total bFGF eluted at 1.0 mol/l NaCl (peak 2) although bFGF continued to be extracted by 1.5 and 2.0 mol/l NaCl. Significantly more bFGF was eluted by graded NaCl extraction than repeated 0.15 mol/l NaCl extraction (mean 78.1 (SEM 10.8) pg/mg NaCl extraction versus 43.5 (5.2) pg/mg control extraction, p<0.05; fig 7A). Heparin and 3.0 mol/l NaCl extracted further bFGF from both NaCl and control extracted mucosa (fig7B). More bFGF was released from control extracted mucosa by heparin and 3.0 mol/l NaCl than NaCl extracted mucosa (fig 7B), presumably because of the limited bFGF extraction with repeated 0.15 mol/l NaCl incubations which had taken place beforehand.

(A) bFGF extraction from antral mucosa of control (non-GU) and gastric ulcer (GU) patients by NaCl. *p<0.05. (B) bFGF extraction from antral mucosa of non-GU and GU patients by 100 μg/ml heparin and 3.0 mol/l NaCl. +p=0.03.
Total bFGF concentrations extracted by graded NaCl and heparin extraction were significantly lower in ulcer patients than non-ulcer controls (74.5 (8.7) pg/mg ulcer versus 109.5 (23.7) pg/mg non-ulcer, p=0.02). During NaCl and heparin elution, bFGF extracted from ulcer patients was consistently less at each molarity than in non-ulcer controls. However, there was no difference in the proportion of total bFGF which eluted at a particular NaCl molarity between ulcer and non-ulcer patients. After control extractions, heparin incubation with non-ulcer mucosa eluted significantly more bFGF than with mucosa from ulcer patients (18.7 (7.9) pg/mg non-ulcer versus 5.8 (1.0) pg/mg ulcer; p=0.03; fig 7B). There was no difference in bFGF extracted by 0.15 mol/l NaCl or total bFGF extracted between those patients with an active ulcer (3/7 taking acid suppressing drugs) and those with a healed ulcer (7/8 taking acid suppressing drugs).
INDUCTION OF bFGF IN GASTRIC ULCERS
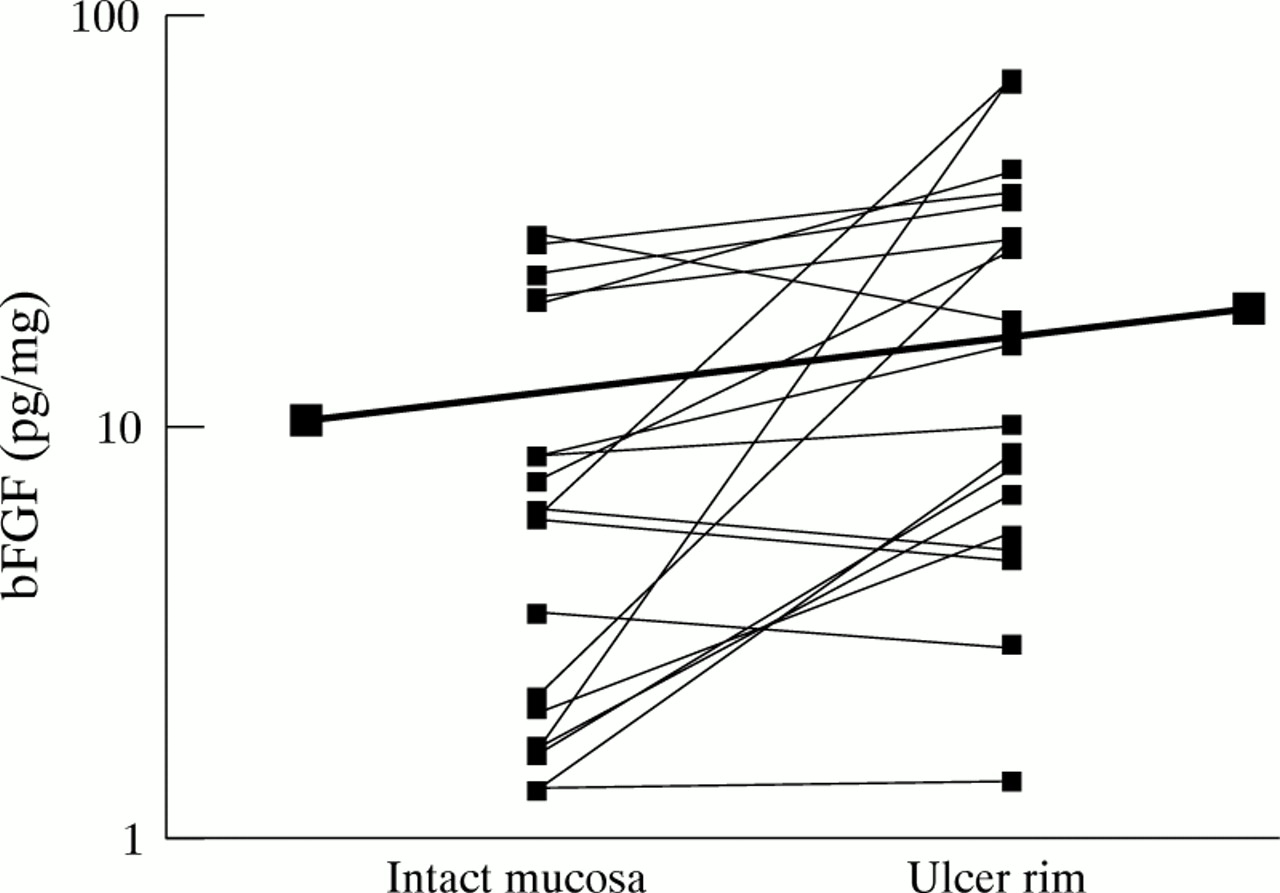
bFGF concentrations were also measured in gastric mucosal homogenate supernatants obtained from the rim of 19 antral gastric ulcers and paired intact antral mucosa from 17 patients at endoscopy (table 1). bFGF concentrations at the ulcer rim were 2.2 (CI 1.2–4.0) fold higher than corresponding values in intact antral mucosa (fig 8). Induction of bFGF at the rim of H pylori negative ulcers (n=11) was greater (3.3× increase (CI 1.2–8.9)) than at the rim ofH pylori positive ulcers (n=8; 1.2× increase (CI 0.8–1.9)). However, the difference in bFGF induction between H pylori positive and negative ulcers just failed to reach statistical significance (p=0.059). Concentrations of bFGF at the rim of non-NSAID associated ulcers (mean 27.6 (CI 0–59.3) pg/mg, n=5; diameter 5–35 mm) were not significantly greater than values at the rim of NSAID associated ulcers (mean 16.2 (5.2–27.2) pg/mg, n=14; diameter 4–20 mm; p=0.6).

Increased bFGF concentrations at the rim of gastric ulcers compared with intact gastric mucosa. The thick line joins the mean values for the two groups. The thin lines join individual pairs of data.
LOCALISATION OF bFGF IN GASTRIC ULCERS
Localisation of bFGF protein and mRNA was investigated in sections from nine chronic gastric ulcers. Mean (SEM) age of patients was 74 (2) years. Three patients were smokers and three patients were taking NSAIDs. Mean (SEM) ulcer diameter recorded in the operation notes was 12.5 (3) mm (n=4). Two ulcers were described as “large”. Ulcer size was not recorded in three cases. Ulcers were located as follows: three antrum, two incisura, four body. All ulcers were on the lesser curve.
Immunohistochemistry
Immunoreactive bFGF was localised to granulation tissue microvessels (fig 9A) and staining was also prominent in the extracellular matrix (ECM) of granulation tissue (fig 9A). A similar distribution of bFGF was seen in granulation tissue of all gastric ulcers examined. In seven of nine ulcers, there was increased immunostaining for bFGF in epithelial cells at the ulcer edge (fig 9B). In contrast to the distribution of bFGF in intact gastric epithelium, bFGF was localised to superficial epithelium including surface epithelial cells at the margin of the ulcer defect (fig 9B). There was also staining of granulation tissue fibroblasts (fig 9C). Muscularis mucosae and deeper smooth muscle fibres showed prominent bFGF staining especially when breached by ulcer granulation tissue (fig 9D). There was no obvious qualitative difference in bFGF immunoreactivity between NSAID and non-NSAID associated gastric ulcers.

Immunohistochemistry for bFGF in gastric ulcers. (A) Granulation tissue. Staining of capillary endothelium (arrow) and extracellular matrix (ECM) (asterisk). Bar=20 μm. (B) Epithelium at the ulcer rim. Increased staining of surface (arrow head) and glandular epithelium compared with intact mucosa. Bar=20 μm. (C) Granulation tissue. Staining of fibroblasts. Scale as for (B). (D) Smooth muscle breached by ulceration. Intense staining of smooth muscle cells. Bar=30 μm.
In situ hybridisation
There was hybridisation of the bFGF probe with bFGF mRNA over granulation tissue microvessel endothelial cells, fibroblasts, and macrophages (fig 10A). There was signal localisation in the muscle layer of arterioles and over smooth muscle fibres surrounded by granulation tissue (fig 10B). There was no evidence of bFGF mRNA localisation in epithelial cells near the ulcer rim despite the immunohistochemical findings.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In situ hybridisation of bFGF mRNA with a 450 bp35S labelled cDNA probe. (A) Gastric ulcer granulation tissue. Signal over mononuclear cells and endothelial cells of a capillary (asterisk). Bar=40 μm. (B) Gastric ulcer. Signal over smooth muscle cells breached by ulcer granulation tissue.
Discussion
In these studies we have characterised bFGF expression in normal human gastric mucosa by a number of techniques including heparin affinity chromatography, ELISA, bioassay, and immunohistochemistry. The size (19 kDa) and elution profile from heparin of gastric mucosal bFGF is in keeping with previous reports of bFGF isolation from several tissues, including a gastric carcinoma resection specimen.4 ,13 ,19-29 In keeping with other studies, there was no evidence of high molecular weight bFGF30 in gastric mucosa when assessed by western blot analysis. The 19 kDa bFGF is likely to be the full 154 amino acid protein predicted by cloning of the human bFGF gene.20 The cocktail of protease inhibitors used during initial protein extraction seems to have prevented the formation of any N-terminal truncated forms of bFGF present in other studies.20 We also used a gastric endothelial cell proliferation assay which confirmed that gastric mucosal bFGF had physiologically relevant bioactivity similar to authentic bFGF.12
The immunohistochemical studies revealed immunoreactive bFGF in endothelial cells, smooth muscle cells, and ECM. These findings are consistent with previous immunohistochemical studies of other human tissues5 ,6 ,29 ,31 and data showing that endothelial cells can synthesise bFGF in vitro.32 Diffuse staining of ECM may represent bFGF sequestered by extracellular HSPGs. Our studies showed patchy staining of epithelial cells; existing data regarding epithelial bFGF expression are conflicting. One study did not detect bFGF in epithelium5 while another showed staining of epithelial basement membranes.6 Others have shown cytoplasmic staining in normal epithelium33 and gastric carcinoma cells.34
Our inability to show bFGF mRNA in normal gastric mucosa by northern blot analysis or in situ hybridisation suggests that we detected stored bFGF which had been synthesised previously. This finding is in keeping with data from a variety of animal tissues which suggest that bFGF mRNA synthesis in normal tissues occurs at a very low level.35 ,36 bFGF is likely to be synthesised by gastric mucosal cells at a low concentration and then stored in association with mucosal HSPGs in an inactive state ready for future mobilisation.9
The most striking finding of our study was the dramatic reduction in intact antral mucosal bFGF concentrations measured by ELISA in gastric ulcer patients. This could not be accounted for by differences between ulcer and non-ulcer patient demographic data and was not explained by NSAID use, H pylori status, or presence of gastritis. We initially used a simple bFGF extraction technique with recovery of bFGF into 0.15 mol/l NaCl. This left open the possibility that the apparent reduction in bFGF concentrations in gastric ulcer patients might have arisen spuriously as a result of more avid binding to mucosa of similar amounts of bFGF (which could also account for increased immunohistochemical staining in these patients). However the NaCl and heparin extraction experiments confirmed that gastric ulcer patients had a true reduction in intact antral mucosal bFGF compared with non-ulcer controls and that there were no alterations in bFGF elution profile to suggest differences in strength of binding of bFGF to gastric mucosal HSPGs. The greater degree of reactive gastritis (and superficial lamina propria muscle bFGF staining) in the small subset of gastric ulcer patients which underwent bFGF histological analysis probably explains the paradoxical increase in immunohistochemical staining despite low bFGF concentrations measurable by ELISA. We have also shown that acid suppressing drugs increase intact mucosal bFGF concentrations. However this effect was smaller in magnitude than the depression in bFGF concentrations seen in gastric ulcer compared with control patients. This is likely to be due to protection of acid labile bFGF from luminal acid.4 Our data are consistent with the only other study in which gastric mucosal bFGF concentrations have been measured in humans by ELISA.37
Approximately one third of extractable mucosal bFGF was released by incubation with 0.15 mol/l NaCl. Basic FGF released in this manner may not be bound to HSPGs and may have been released by mechanical trauma alone. Alternatively, a fraction of bFGF bound to HSPGs could have been released by mucosal heparitinases activated by homogenisation. Further mucosal bFGF was released by increasing NaCl molarity which peaked at 1.0 mol/l NaCl. The very low concentrations of bFGF extracted by control elution confirmed that release of bFGF was related to increasing NaCl molarity. The profile of bFGF elution from gastric mucosa reported here is consistent with reports in the literature by workers who have investigated elution of HSPGs from immobilised bFGF9 ,38 ,39 and is further evidence that HSPGs bind bFGF in human gastric mucosa. Thus, there appears to be two pools of gastric mucosal bFGF: “free” bFGF which is not bound to HSPGs and which can be released by mechanical perturbation; and “bound” bFGF which is bound to HSPGs and is eluted by 0.5–3.0 mol/l NaCl and heparin. Increased extraction of bFGF from mucosa of non-ulcer patients by heparin would suggest that the excess bFGF in these patients is “bound” bFGF. The absolute bFGF concentrations extracted by 0.15 mol/l NaCl during the NaCl and heparin extraction experiments were lower than those obtained in the initial extraction experiments. This could be explained by degradation of bFGF during the 10 minute incubation period before centrifugation or by absorption of bFGF during gel filtration in the extraction experiments.
Reduced mucosal bFGF content could be explained by decreased bFGF synthesis, increased bFGF degradation, or both. Even if bFGF synthesis in intact gastric mucosa is very low, reduced bFGF synthesis over a long period of time could account for reduced mucosal bFGF content if the bFGF degradation rate remained constant. Increased degradation of mucosal bFGF is more likely to account for reduced mucosal bFGF concentrations. As bFGF is very acid labile, back diffusion of luminal acid into superficial mucosa could destroy significant amounts of mucosal bFGF. It has been calculated that net flux of H+from lumen to mucosa occurs in the normal human stomach (1–2 mEq/15 minutes) which is increased after instillation of an acid load.40 ,41 Increased acid back diffusion has previously been noted in gastric ulcer patients before and after healing.42 Although H pylori and NSAIDs have the potential to increase acid back diffusion, our studies did not identify either as a significant factor in determining mucosal bFGF concentrations.
The presence of a fibroblast growth factor binding protein (FGF-BP) in human gastric mucosa could provide an alternative explanation for the ELISA and immunohistochemistry data obtained in this study. A secreted FGF-BP, which mobilises and activates locally stored bFGF in vitro,43 has recently been shown to play an important role in squamous cell carcinoma growth and angiogenesis.44 If FGF-BP binding to bFGF in gastric mucosa involved the bFGF epitopes which are recognised by Mab52 and Mab98, then altered expression of FGF-BP could account for the differences in measurable bFGF concentrations between gastric ulcer patients and controls and also the paradoxical ELISA and immunohistochemistry data.
Against a background of low bFGF concentrations, expression of bFGF was increased in gastric ulcers. Our immunohistochemistry and in situ hybridisation studies showed bFGF expression by microvessel endothelial cells, mononuclear cells, and fibroblasts in granulation tissue. Capillary endothelial cells and macrophages have previously been shown to synthesise bFGF in culture32 ,45 and bFGF has previously been noted in granulation tissue fibroblasts and macrophages in gastric ulcers in animals1 ,46 and humans.47 bFGF immunoreactivity was prominent in muscle especially if this layer was breached by an ulcer. Muscle fibres could act as an intracellular store of bFGF for release if tissue injury becomes deep enough to cause damage to this layer. We showed bFGF protein in epithelial cells at the rim of ulcers. Induction of bFGF synthesis may be analogous to up regulation of EGF synthesis at the edge of chronic ulcers.48 However, increased bFGF mRNA could not be detected in epithelial cells by in situ hybridisation. This discrepancy suggests that induction of bFGF mRNA synthesis may be an early event after gastric mucosal injury, as has been reported in rats49 and that the peak of bFGF synthesis was missed in the surgically resected gastric ulcers examined in this study. Further studies of bFGF mRNA expression in gastric ulcers by reverse transcriptase polymerase chain reaction (RT-PCR) and in situ PCR are required to determine whether there is an increase in bFGF mRNA synthesis in epithelial cells at the ulcer rim. Alternatively, epithelial cells could have taken up bFGF released from ECM after digestion of HSPGs by granulation tissue heparanases. This hypothesis could be tested by incubation of isolated gastric epithelial cells with exogenous bFGF followed by demonstration of intracellular accumulation of bFGF by subcellular fractionation techniques. Epithelial cell bFGF could play a role in angiogenesis in adjacent granulation tissue or have actions on ulcer rim epithelium. There is evidence that bFGF promotes frog gastric epithelial cell restitution in vitro50 and restitution of rat intestinal epithelial cells in culture.51
The increase in bFGF concentrations at the ulcer rim was smaller than the reduction of bFGF in intact mucosa compared with non-ulcer patients. These data are also consistent with the study by Tsujiet al.37
There was a trend towards decreased ulcer rim bFGF concentrations inH pylori positive gastric ulcers compared with H pylori negative ulcers. This was not explained by differences in NSAID use between the two groups. The lack of statistical significance could have been caused by type II error and more ulcers need to be studied. Two mechanisms by which the presence of H pyloricould reduce bFGF concentrations could be implicated: H pylori lysates contain protease activity which reduces bFGF induced mitogenic activity52; and H pyloriexpresses cell surface HSPGs which bind bFGF avidly and could sequester mucosal bFGF.53 Decreased induction of bFGF in NSAID associated gastric ulcers was not shown and does not account for the impaired angiogenic response in such ulcers.54
In summary, bFGF is present in normal human gastric mucosa and its expression is up regulated in gastric ulcers. Gastric ulcer patients appear to have a constitutional deficiency of mucosal bFGF which is not explained by altered binding by gastric mucosal HSPGs. Further experiments are required to test the hypothesis that reduced mucosal bFGF content predisposes to ulcer formation.
Acknowledgments
J L Brough was supported by a grant from the Trent Research Scheme. Part of this work has been presented to the American Gastroenterological Association in abstract form (Gastroenterology 1994;106:A97;Gastroenterology 1995;108:A117). The authors wish to thank Professor John T Gallagher, Christie Hospital, Manchester, for his advice regarding the bFGF extraction experiments.