Article Text

Abstract
Background: The hepatitis C virus (HCV) mutates within human leucocyte antigen (HLA) class I restricted immunodominant epitopes of the non-structural (NS) 3/4A protease to escape cytotoxic T lymphocyte (CTL) recognition and promote viral persistence. However, variability is not unlimited, and sometimes almost absent, and factors that restrict viral variability have not been defined experimentally.
Aims: We wished to explore whether the variability of the immunodominant CTL epitope at residues 1073–1081 of the NS3 protease was limited by viral fitness.
Patients: Venous blood was obtained from six patients (four HLA-A2+) with chronic HCV infection and from one HLA-A2+ patient with acute HCV infection.
Methods: NS3/4A genes were amplified from serum, cloned in a eukaryotic expression plasmid, sequenced, and expressed. CTL recognition of naturally occurring and artificially introduced escape mutations in HLA-A2-restricted NS3 epitopes were determined using CTLs from human blood and genetically immunised HLA-A2-transgenic mice. HCV replicons were used to test the effect of escape mutations on HCV protease activity and RNA replication.
Results: Sequence analysis of NS3/4A confirmed low genetic variability. The major viral species had functional proteases with 1073–1081 epitopes that were generally recognised by cross reactive human and murine HLA-A2 restricted CTLs. Introduction of mutations at five positions of the 1073–1081 epitope prevented CTL recognition but three of these reduced protease activity and RNA replication.
Conclusions: Viral fitness can indeed limit the variability of HCV within immunological epitopes. This helps to explain why certain immunological escape variants never appear as a major viral species in infected humans.
- HCV, hepatitis C virus
- HBV, hepatitis B virus
- HLA, human leucocyte antigen
- NS, non-structural
- CTL, cytotoxic T lymphocyte
- PBMC, peripheral blood mononuclear cells
- SFV, semliki forest virus
- PCR, polymerase chain reaction
- DMEM, Dulbecco’s modified Eagle’s medium
- IFN-γ, interferon γ
- hepatitis C virus
- NS3
- immune escape
- cytotoxic T lymphocyte
- viral fitness
- replicon
Statistics from Altmetric.com
- HCV, hepatitis C virus
- HBV, hepatitis B virus
- HLA, human leucocyte antigen
- NS, non-structural
- CTL, cytotoxic T lymphocyte
- PBMC, peripheral blood mononuclear cells
- SFV, semliki forest virus
- PCR, polymerase chain reaction
- DMEM, Dulbecco’s modified Eagle’s medium
- IFN-γ, interferon γ
The non-lytic hepatitis C virus (HCV) is a leading cause of chronic liver disease worldwide. The killing of infected cells by cytotoxic T lymphocytes (CTLs) is of crucial importance as a general mechanism in clearing non-lytic viral infections.1,2 Indeed, a strong and maintained CTL response to various HCV proteins has been found to correlate with viral clearance.3–8 If these CTL responses become functionally impaired, patients may progress to chronic infection.4,9–13
Hallmarks of HCV infection are its high propensity in establishing persistence and the fact that the viral RNA genome undergoes extensive mutations during replication. This allows for the evolution of multiple viral species in a single patient.14 Hence one way for HCV to ensure viral persistence may be to use the high genetic variability and accumulate mutations within CTL epitopes to impair CTL recognition.6,15–18 Additional mechanisms favouring persistence are viral proteins that directly inhibit cellular interferon signalling,19 functional impairment of CTLs,20,21 or mutations that prevent antigen processing and presentation.22 Which of these escape mechanisms are the most important in infected humans is not known.
Although immune escape by mutations within CTL epitopes has been convincingly proposed as a key mechanism in chronic HCV infection, some epitopes do not seem to mutate extensively. Possibly the best example of a CTL epitope with a very low variability is the immunodominant human leucocyte antigen (HLA)-A2 restricted epitope at residues 1073–1081 within the non-structural (NS)3 protease.22,23 Although this epitope rarely changes, CTLs specific for the 1073–1081 NS3 epitope are of key importance and have been correlated with clearance of infection.3 The conserved nature of the NS3 gene has been attributed to the fact that this protein encompasses both protease and NTPase dependent helicase activities.24,25 This indirectly implies that immune escape mutations within these enzymes may be lethal for the virus. Regarding the human immunodeficiency virus type 1, it is well known that immune escape mutations may appear at the expense of viral fitness.26–28 Thus it is possible that mutations mediating immune escape within the HCV protease negatively affect viral fitness, thereby explaining the conserved nature of some immunodominant epitopes within the HCV protease. This hypothesis was tested in the present study.
MATERIALS AND METHODS
Serum samples
Venous blood samples were obtained in heparin tubes from five patients with chronic HCV infection of genotype 1 (four males and one female; aged 27–61 years) with detectable HCV-RNA (Roche Amplicor, Roche Raritran, New Jersey, USA). From two patients, two samples from each patient were obtained 4–6 months apart. Plasma aliquots were kept at −80°C until use.
Peripheral blood mononuclear cells (PBMC) were stained with an FITC conjugated anti-HLA-A2 antibody (R&D Systems, Minneapolis, Minnesota, USA) and the presence of the HLA-A2 molecule was determined by flow cytometry.
PBMC were also obtained from three HLA-A2 positive patients: one patients had cleared an acute HCV infection six months prior to sampling; one had cleared a chronic HCV genotype 1b infection prior to sampling (earlier samples included in the sequence analysis as patient No 1); and one patient was chronically infected by HCV genotype 1a. All sampling and analysis of human samples was approved by the local ethics committee.
Mice and immunisations
Inbred C57BL/6 (H-2b) mice were obtained from a commercial vendor (Möllegard, Denmark). Inbred HHD (HHD+ H-2Db−/− β2m−/−) mice transgenic for HLA-A2.1 monochain histocompatibility class I molecule were kindly provided by Dr F Lemonnier (Institut Pasteur, France).29 Six to 12 week old mice were used. The animal ethics committee approved all experimental protocols.
Groups of 3–7 mice were immunised with 100 μg of plasmid DNA suspended in phosphate buffered saline given by needle injections in the tibialis anterior muscle. Gene gun based immunisations were performed using 4 µg of plasmid DNA as described.30,31 Alternatively, mice were immunised subcutaneously with 1×107 semliki forest virus (SFV) particles in 100 μl of phosphate buffered saline.30,31 All groups were given the same dose twice, four weeks apart.
Peptide immunisation was performed by subcutaneous immunisation in the base of the tail with 100 μg of peptide and 140 μg of hepatitis B virus (HBV) core helper epitope 128–140 peptide mixed 1:1 with incomplete Freund’s adjuvant.30
Plasmids and cloning of HCV NS3/4A genes
Cloning and codon optimisation of a full length NS3/4A gene has been reported previously,30,31 and the coNS3/4A gene was cloned in the SFV vector system using the same approach as described previously.30,31
Full length NS3/4A genes were isolated and cloned from human serum by the polymerase chain reaction (PCR), as previously described.31 In brief, total serum RNA was extracted from 200 µl of serum (QIAamp Viral RNA kit; Qiagen, Crawley, UK), and complementary DNA was generated as described previously.31 HCV NS3/4A fragments were amplified by a nested PCR using a Taq polymerase with proof reading ability (Expand High Fidelity PCR System; Roche).31 PCR products were separated by gel electrophoresis and visualised under UV light. Amplicons were cloned into the pVAX1 plasmid (Invitrogen GmbH, Karlsruhe, Germany) using EcoRI and XbaI (Roche) sites.31 NS3/4A containing plasmids were identified, grown, and purified as described in detail previously.31 All clones were sequenced by Genera dB (Riga, Latvia).
Generation of pFK-PI-lucEI/NS3-3′ plasmids has been described previously.32 The plasmid pFK-PI-lucEI/NS3-3′/ET carrying cell culture adaptive mutations in NS3 and in NS4B, and the pTM-NS3-3′ plasmid, have been described elsewhere.33,34
Synthetic peptides
Peptides were produced corresponding to HLA-A2 epitopes of the CMV pp65 protein35 (sequence NLVPMVATV) or the NS3 region of HCV36,37 (fig 1). Peptides corresponding to a murine HBV core helper epitope 128–140 (TPPATRPPNAPIL38) and a murine H-2b restricted CTL epitope (GAVQNEVTL31) were also used. Peptides were kindly synthesised by Dr Michael Levi (Tripep, Stockholm, Sweden) using an automated synthesiser.39,40

Sequence variants of HLA-A2 restricted epitopes found within the NS3/4A gene among 48 full length clones from five patients with chronic hepatitis C virus infections. Sequences in italic represent Genebank sequences. HLA, human leucocyte antigen; CTL, cytotoxic T lymphocyte.
In vitro mutagenesis
Mutant plasmids in which each residue within the 1073–1081 (CINGVCWTV) epitope was sequentially replaced by alanine were generated by site directed in vitro mutagenesis (Quick-Change, Site-Directed Mutagenesis Kit; Stratagene, La Jolla, California, USA) using primers containing the specific mutation.31 The mutant sequences were confirmed by sequencing.
In vitro translation assay
The functionality of the NS3/4A protease was analysed by an in vitro transcription and translation assay (TNT; Promega, Madison, Wisconsin, USA), as described previously.31,41 A functional NS3 protease generates two protein bands on a sodium dodecyl sulphate-polyacrylamide gel, a free NS3 protein, and an NS3-NS4A fusion protein, whereas a non-functional protease only generates the NS3-NS4A fusion protein band.
Cell lines
RMA-S cells (H-2b) and RMAS-HHD (HHD+ H-2b−; kindly provided by Dr F Lemonnier, Institut Pasteur, France) were maintained in RPMI 1640 medium supplemented with 5% fetal calf serum and 800 μg/ml G418. Monolayers of the human hepatoma cell line Huh-7 were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen) supplemented with non-essential amino acids and 10% fetal calf serum (complete DMEM). All media also contained 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin, and cells were grown in a humidified incubator at +37°C and with 5% CO2.
Peptide stabilisation assay and detection of NS3/4A specific CTL
The ability of NS3/4A derived HLA-A2 peptides to bind to HHD molecules was determined in an RMAS-HHD stabilisation assay.31,42,43
NS3/4A specific CTLs were detected as described previously.30,31 In brief, five day restimulation cultures were set with 20×106 splenocytes from immunised mice and an equal amount of irradiated (2000 rad) syngeneic splenocytes, and 0.05 μM of peptide. CTLs were detected by a four hour 51Cr release assay using peptide pulsed RMAS-HHD target cells.
Human CTLs were detected by NS3-peptide induced interferon γ (IFN-γ) production using a commercially available ELISPOT assay (human IFN-γ ELISPOT Set; Becton Dickinson, Franklin Lakes, New Jersey, USA). Human PBMCs were incubated with NS3 derived HLA-A2 peptides at a concentration of 10 µM (saturating conditions) to ensure detection of low avidity interactions.
In vitro transcription and transient HCV replication assay
In vitro transcripts were generated from the pFK-PI-lucEI/NS3-3′ plasmid and electroporation was performed as described previously.33,44,45 In brief, a total of 5 µg of in vitro transcribed RNA were mixed with 400 µl suspensions of 107 highly permissive Huh-7 cells per ml46 and, after electroporation, were diluted in 20 ml of complete DMEM. Aliquots (2 ml) were seeded onto six well plates and harvested 4, 24, 48, 72, and 96 hours later. Cells were lysed and after addition of luciferin solution, activity was measured in a luminometer (Lumat LB9507; Berthold, Freiburg, Germany) for 20 seconds. All luciferase assays were performed in duplicate.
Metabolic radiolabelling of proteins and immunoprecipitation
Metabolic radiolabelling of proteins and immunoprecipitation was performed essentially as described previously.47 In brief, a total of 7×105 Huh-7/T7 cells44 were transfected with 8 µg of T7 based expression constructs carrying the NS3 to NS5B coding region downstream of the EMCV-IRES (Lipofectamine 2000; Invitrogen). Cells were then starved using methionine free medium for 30 minutes and then metabolically labelled with 300 µCi [35S]-methionine (NEN Life Science Products, Cologne, Germany). Cells were washed and lysed and lysates were clarified in a microfuge. Cell lysates were divided into four aliquots and immunoprecipitated with protein A agarose (BioRad Laboratories GmbH, Munich, Germany) loaded with HCV specific antisera. Immunocomplexes were washed, resuspended, denatured, and proteins were analysed by 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis.47
RESULTS
Genetic variability of HCV NS3/4A
We amplified and cloned 48 full length HCV NS3/4A genes from seven serum samples obtained from five patients with chronic HCV infection (fig 1). Sequence analysis confirmed that the genetic variability within the NS3/4A genes was limited. The number of polymorphic sites among 141 nucleotides encoding five known HLA-A2 epitopes23,48–50 did not differ between the 25 clones from the patients with, or the 23 clones from the patients without, the HLA-A2 allele. The number of synonymous and non-synonymous mutations was the same inside or outside the HLA-A2 restricted epitopes in both HLA-A2 positive and negative patients (data not shown).
HLA-A2 binding and CTL recognition of NS3 derived HLA-A2 restricted epitopes
We next tested the binding avidity to HLA-A2 molecules and recognition of natural and mutant peptides corresponding to HLA-A2 epitopes by human and murine CTLs. Binding avidity was tested by stabilisation of expression of HLA-A2 molecules on RMAS-HHD cells by adding dilutions (0.001 to 100 µM) of HLA-A2 peptides. CTL recognition was tested by adding a saturating concentration (10 µM) to also allow for binding and recognition of low avidity HLA-A2 epitope-peptides in a standard toxicity assay.
Four of five naturally occurring HLA-A2 restricted epitope variants among our 48 NS3/4A clones bound HLA-A2 molecules with varying efficiency (fig 2A). Of the naturally occurring variants of the immunodominant 1073-1081 epitope, only the 1081Val to 1081Ala substitution reduced HLA-A2 binding by >100-fold (fig 2A). Such a mutation may impair antigen presentation and is consistent with the previously reported recognition by CTLs, albeit with a >50-fold reduced binding avidity of the 1081Ala substitution peptide.23

Binding of the naturally occurring (A) and mutant (B) NS3 derived HLA-A2 restricted peptides to HLA-A2 transfected RMA-S cells (A, B). Values are given as the lowest peptide dilution inducing positive fluorescence. Low peptide concentrations indicate higher HLA-A2 binding avidity. Broken vertical lines in (B) indicate values that are 100-fold higher or lower than the parental peptide. Also shown are the recognition 1073–1081 peptide analogues by human (C–E) and murine (F) HLA-A2 restricted cytotoxic T lymphocytes (CTLs), as determined by EPISPOT (C–E) or by lysis of radioactively labelled peptide loaded HLA-A2 transfected RMA-S cells (F). Data are given as the number of interferon γ (IFN-γ) producing cells per 106 peripheral blood mononuclear cells (PBMC) (C–E) or per cent specific lysis (F). Broken vertical lines in (C–E) indicate values 50% of the original peptide. Pat, patient; gt, genotype; HCV, hepatitis C virus.
Natural variations of the 1169–1177 epitope did not alter HLA-A2 binding whereas the 1287–1296 epitope peptides bound poorly to HLA-A2 (fig 2A). Natural variants of the epitope 1406–1415 showed a broader range of HLA-A2 binding avidities (fig 2A). The natural peptide with the highest binding avidity (KLVALGVNAV; fig 2A) was found in 10 of 10 clones from the HLA-A2+ patient No 2 (fig 1). In the HLA-A2+ patient No 3 infected by HCV genotype 1b, the avidity reducing 1411Gly to 1411Arg mutation appeared in one clone in a second sample (fig 1, 2A), possibly representing immune escape. However, the same mutation was not found when performing sequencing of a later sample from this patient (data not shown). It has been shown that a 1410Leu to 1410Met mutation may represent immune escape.51 No variability was present within the epitope at residues 1590–1598 among our NS3/4A sequences although a 1598Val to 1598Gln substitution was found in Genebank (fig 2A). Thus HLA-A2 epitopes representing the major viral species of naturally occurring variants all bound HLA-A2. Immune escape variants with reduced HLA-A2 binding avidities were only found among minor viral species.
HLA-A2 binding of the immunodominant 1073–1081 epitope was analysed in detail using alanine substitution peptide analogues. Alanine at positions 1074, 1079, and 1081 reduced binding by 100- to 1000-fold, indicating that these interact with the HLA-A2 molecule (agretopic; fig 2B). Thus such mutations impair HLA-A2 binding and antigen presentation.
We next tested if naturally occurring and mutant 1073–1081 epitope peptides were recognised by HLA-A2 restricted CTLs from one patient who successfully cleared an acute HCV infection, one who was cured from a chronic infection by therapy (patient No 1), and one with a chronic HCV infection (fig 2C–E). The HLA-A2 restricted CTL response in the patient with acute infection and the patient who cleared a chronic HCV infection focused on the 1073–1081-epitope (fig 2C, D). In the acute patient, alanine substitutions at residues 1075, 1077, and 1079 impaired CTL recognition, suggesting that residues 1075 and 1077 interact with the T cell receptor (epitopic). Interestingly, the CTLs in this patient were poorly cross reactive between the highly conservative Ile to Val substitution (fig 2C). In contrast, CTLs from the patient who was cured from a chronic HCV genotype 1b infection were cross reactive between Ile (genotype 1a) and Val (genotype 1b) containing peptides, although the genotype 1b peptide was slightly better recognised that the 1a peptide (fig 2D). In the patient with an ongoing chronic HCV genotype 1a infection, CTLs were equally sensitive to mutations as CTLs from patients with acute HCV infection (fig 2E).
Recognition patterns of CTLs from NS3/4A DNA immunised HLA-A2 transgenic mice revealed that alanine substitutions of positions 1075, 1077, and 1079 impaired recognition (fig 2F). Also, it is interesting to note that the peptide with an Ala at residue 1081 still bound the HLA-A2 molecule and was recognised, albeit weakly, by both human (fig 2C, E) and murine (fig 2F) CTLs. Overall, human and murine recognition of the tested NS3 derived HLA-A2 epitopes in general and the 1073–1081 epitope in particular seemed to be comparable.
In conclusion, residues 1074, 1079, and 1081 are agretopic residues (binding to the HLA-A2 molecule) whereas residues 1075, 1077, and 1079 are epitopic residues (interacting with the T cell receptor). Thus mutations at these five positions should help the virus to escape the immunodominant host CTL response.
Immunodominance among HLA-A2 restricted CTL epitopes within NS3
As recognition of the 1073–1081 epitope was fully comparable between humans and HLA-A2-Tg mice, we evaluated the immunogenicity and immunodominance of the NS3/4A protease in HLA-A2-Tg mice. The NS3/4A gene primed a CTL response that was focused on the 1073–1081 epitope (fig 3A). CTLs primed by the CINGVCWTV 1073–1081 epitope sequence of the vaccine were cross reactive between peptides corresponding to genotype 1a and 1b specific peptides regardless of the immunogen (fig 3). Thus the fine specificity in the HLA-A2-Tg mice resembles some, but not all, of the human HLA-A2-restricted CTLs. As is expected from binding studies, the peptide with the 1081Val to 1081Ala substitution was not recognised, confirming that this represents an immune escape variant (fig 3). HLA-A2-Tg mice immunised with an NS3/4A gene with the 1073–1081 epitope sequence CINGVCWSV, derived from patient No 4 (sequence 4:1, fig 1) primed the same type of response as the original NS3/4A gene (fig 3A–D, 3Q–T).

Immunodominance among HLA-A2 restricted NS3 derived cytotoxic T lymphocyte (CTL) epitopes in HLA-A2-Tg mice genetically immunised intramuscularly (im) with a wild-type (wt) NS3/4A coding plasmid (A–D), using a gene gun with a codon optimised NS3/4A coding plasmid (E–H), subcutaneously (sc) with wild-type NS3/4A coding semliki forest virus (SFV) particles (I–L), subcutaneously with a codon optimised NS3/4A coding SFV particles (M–P), intramuscularly with an NS3/4A plasmid obtained from patient No 4:1 (Q–T), or intramuscularly with an NS3/4A coding plasmid with Ala mutations silencing the 1073–1081 epitope (U–Y). The epitope sequences of the immunogen used in (A–P) are given on top. The insert in (U) shows intramuscular immunisation of wild-type C57BL/6 mice with the same plasmid as in (Q). Results are given as mean per cent specific lysis.
To exclude the possibility that the immunodominance of the 1073–1081 epitope was caused by the DNA based immunisation, we immunised HLA-A2-Tg mice using the SFV vector. SFV and HCV have positive sense single stranded RNA genomes and generate high levels of double stranded (ds) RNA intermediates,52 which may influence CTL priming.53 However, the CTL response did not change when using the SFV vectors (fig 3I–P). Thus endogenously produced HCV NS3/4A protease, regardless of whether expressed from a DNA plasmid or RNA vector, preferentially primes 1073–1081 specific CTLs in a HLA-A2 positive host. This is consistent with the immunodominance of the 1073–1081 HLA-A2 restricted epitope in HCV infected humans (fig 2C, D).3,49
To confirm that the agretopic 1074Ala and 1081Ala mutations mediate immune escape by impaired epitope presentation, a DNA plasmid was generated in which these Ala mutations were introduced. Indeed, immunisation of HLA-A2 Tg mice using this plasmid failed to prime a detectable CTL response to the 1073–1081 epitope (fig 3U–Y). The mutant NS3/4A gene had an intact overall immunogenicity, as wild-type H-2b mice immunised with the mutant plasmid developed a CTL response against the H-2b restricted epitope (fig 3U, insert). Thus mutations that reduce HLA-A2 binding of the 1073–1081 epitope mediates immune escape.
Effect of naturally occurring and targeted mutations within NS3/4A on protease activity
To address whether mutations within the immunodominant 1073–1081 epitope affect viral fitness, we first analysed the functionality of the 48 naturally occurring HCV NS3/4A protease variants. Of the 48 naturally occurring NS3/4A genes tested, four represented variant 1073–1081 epitope sequences (fig 1) and all of these had functional proteases by an in vitro transcription and translation assay (data not shown).
All residues within 1073–1081 of an NS3/4A gene expressing a functional protease were sequentially replaced by alanine to investigate the effect of these on protease activity. We found that alanine substitutions at residues 1074 and 1079 (and possibly 1076) severely impaired protease activity (fig 4). To better understand which degree of variability can be accepted, additional mutations were introduced. The 1074 Ile could be replaced by a Met or a Leu, but not by a Glu or a Lys, which destroyed protease activity (table 1). The 1079 Trp could be substituted by a Phe or Tyr residue, whereas a Met or a His substitution impaired cleavage at the NS3/4A junction (data not shown). Thus conservative changes at residues 1074 and 1079 clearly affect protease activity.
Polyprotein processing and replicative capacity of hepatitis C virus (HCV) replicons carrying mutations within the 1073–1081 epitope

Analysis of the activities of various NS3/4A proteases by an in vitro transcription and translation assay. Protease activity was tested by an in vitro transcription and translation assay of NS3/4A genes with wild type (wt) or mutant 1073–1081 epitope sequences. The upper band corresponds to the non-cleaved NS3/4A fusion protein and the lower band corresponds to the free NS3 protein. A single NS3/4A fusion band represents a non-functional protease.
Polyprotein processing and replicative capacity of HCV replicons with alanine substitutions within the immunodominant epitope
To test whether epitope mutations affected HCV polyprotein processing, the alanine substitutions at positions 1074 and 1081 representing HLA-A2 binding residues were introduced into an expression plasmid encoding the complete NS3 to NS5B region. The 1074Ile to Ala substitution, but not the 1081Val to Ala substitution, modified NS3/4A mediated processing, evidenced by accumulation of an uncleaved NS3/4A and other processing intermediates (fig 5A, lanes 3, 5, 8, and 10). As the amounts of fully processed NS4B, NS5A, and NS5B generated by the single and double mutants were comparable with those of the wild-type, the primary defect was improper cleavage at the NS3/4A junction. Also, the amount of hyperphosphorylated NS5A that migrates slightly faster compared with basal phosphorylated NS5A appeared to be reduced by the 1074Ile to Ala substitution (fig 5A; compare lanes 13 and 14). Additional 1073–1081 epitope mutants revealed that the 1079Trp to Ala substitution impaired processing (table 1). Conservative mutations such as from 1074Ile to 1074Leu or 1074Met had retained processing whereas substitutions by 1074Glu or 1074Lys destroyed the same (table 1). Again, minor changes may at some positions impair protease function.
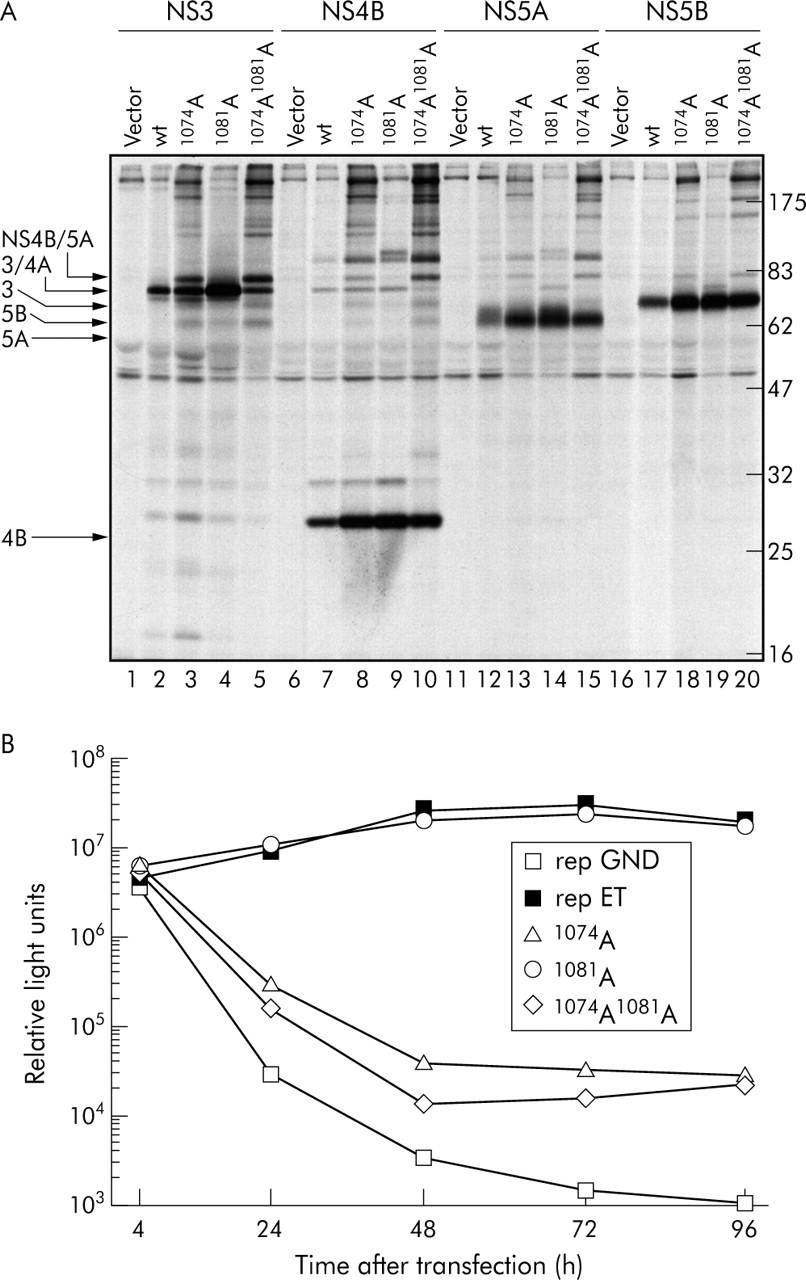
Analysis of the activities of various NS3/4A proteases by polyprotein expression in transfected cells (A) and replication of viral RNA (B). (A) Analysis of polyprotein processing using NS3 to NS5B constructs containing given 1073–1081 epitope mutations. Huh-7/T7 cells were transfected with the control plasmid, wild type (wt) construct, or plasmids containing the 1073–1081 epitope mutants. Four hours later proteins were radiolabelled metabolically with [35S] methionine. Hepatitis C virus specific proteins were isolated from cell lysates by immunoprecipitation using given antisera and proteins were detected after sodium dodecyl sulphate-polyacrylamide gel electrophoresis by autoradiography. (B) Transient replication of replicons carrying the luciferase reporter gene and given mutations in the NS3 1073–1081 epitope. Highly permissive Huh-7 cells were transfected with 5 µg of replicon RNA, and luciferase activity was determined in cell lysates 4, 24, 48, 72, and 96 hours after transfection. RNA transfections and luciferase assays were performed in duplicate.
Next we introduced these NS3 protease mutations into a subgenomic HCV reporter replicon where the amount of expressed luciferase accurately reflects the level of replicon RNA replication.32,54 The replicon containing the 1074Ala substitution had severely impaired replication fitness as luciferase values were reduced about 1000-fold compared with the wild-type (fig 5B). The same was true for the 1074/1081Ala double mutant (fig 5B) whereas the 1081Ala single mutation had no significant effect on RNA replication. Replicons harbouring 1074Glu, 1074Lys, 1075Ala, 1076Ala, or 1079Ala substitutions had severely impaired RNA replication (table 1). In addition, we tested the effect on protease activity of the 1082Tyr to 1082Phe mutation residing C terminal of the immunodominant epitope, which has been found to prevent antigen presentation.22 The 1082Tyr to 1082Phe mutant had intact replication whereas a 1082Ala substitution at the same position impaired replication (table 1). As shown in the summary in fig 6, we conclude that mutations at residues 1074, 1075, 1076, and 1079 of the immunodominant 1073–1081 epitope reduce viral fitness.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Summary cartoon of the immune recognition and mutational analysis of the NS3 derived HLA-A2 restricted cytotoxic T lymphocyte (CTL) epitope at residues 1073–1081. TCR, T cell receptor.
DISCUSSION
A well established dogma is that HCV actively evades the host CTL response. This is done either by accumulation of mutations within CTL epitopes6,15–18,55 or at flanking residues to prevent processing and presentation of the antigenic peptide.22 Selection of such immune escape variants requires a selective pressure by the host CTL response. However, the limited variability within and around the immunodominant HLA-A2 restricted CTL epitope of the NS3 protease is inconsistent with the presence of a selective immunological pressure. However, variability may be limited by viral fitness.
We tested this concept using the immunodominant epitope at residues 1073–1081 within the HCV NS3 protease. Several studies have confirmed the immunodominance of this CTL epitope in HLA-A2+ patients and provided evidence for a role for CTLs to this epitope in the control of infection.3,49 Sequence analysis revealed that the naturally occurring variability within this epitope was limited, consistent with previous observations.22
Naturally occurring peptide epitope variants representing major viral species all bound to HLA-A2, albeit single clones were found that harboured HLA-A2 epitopes with much reduced binding, implying immune escape. Within one immunodominant HLA-A2 epitope of NS3 we found that residues 1074, 1079, and 1081 were responsible for binding to the HLA-A2 molecule. Interestingly, despite the fact that mutations at these positions impaired antigen presentation and therefore effectively mediated immune escape, such variants never appeared as a major viral species. This could suggest that the epitope is not immunodominant or that something prevents the appearance of such mutations. We and others have found that both human and murine HLA-A2 restricted CTLs were indeed to a large extent focused on the 1073–1081 epitope and that they may be cross reactive between the different major viral species. Thus there is no doubt that epitope 1073–1081 is a target for the host CTL response.
The immunodominance of the 1073–1081 specific CTL response was studied in some detail using HLA-A2 transgenic mice. Although one concern may be that CTL recognition may differ between HLA-A2 positive humans and mice, we found that the recognition pattern of murine NS3/4A specific CTLs was comparable with human CTLs. The 1073–1081 epitope was immunodominant in HLA-A2-Tg mice regardless of the immunogen or the delivery route, and silencing of the 1073–1081 epitope through alanine substitutions at residues 1074 and 1081 severely impaired priming of HLA-A2 restricted CTLs. Thus there is no doubt that some non-conservative mutations within the 1073–1081 epitope mediate immune escape.
Viral escape mutation strategies that abolish peptide binding to various human class I alleles have been demonstrated.56,57 However, we could not find evidence that such mutations appear in HLA-A2 positive HCV infected subjects. Thus although the 1073–1081 NS3 CTL epitope is immunodominant and highly immunogenic, and non-conservative mutations severely impair an NS3 specific HLA-A2 restricted response, these mutations never become positively selected. This suggests that these mutations may affect viral fitness.
We showed that by replacing each of the natural 1073–1081 epitope residues of an HCV replicon by alanine, that the residues 1074, 1075, 1076, and 1079 were indeed important for viral RNA replication. Thus mutations at these four positions within the nine amino acid immunodominant 1073–1081 epitope had profound effects on viral fitness. This may help to explain why these mutations are so rare in nature. However, it is possible that viruses with mutations that reduce viral replication may acquire compensatory mutations that restore viral fitness. This can be studied using the newly developed infectious HCV replicon.58
In conclusion, mutations at residues 1074, 1075, 1077, 1079, and 1081 mediated immune escape as these residues interacted either with the HLA molecule or the T cell receptor. However, immune escape at residues 1074, 1075, and 1079 is not likely to appear in nature as these coincide with mutations that reduce the replication of viral RNA, and hence viral fitness. This is the first example showing that the ability of HCV to mutate within immunological epitopes is restricted by effects on viral fitness.
Acknowledgments
The study was supported by the Swedish Science Council grant K2002-16X-09494-12B, the Swedish Cancer Foundation grant 3825-B04-09XCC, the European commission grants QLK2-1999-00588, and activities of the VIRGIL European Network of Excellence on Antiviral Drug Resistance supported by a grant (LSHM-CT-2004-503359) from the Priority 1 “Life Sciences, Genomics and Biotechnology for Health” programme in the 6th Framework Programme of the EU. Dr Lars Frelin was supported by grants from Eirs 50 years Foundation, Gålö Foundation, and Gemzeús Foundation.
REFERENCES
Footnotes
-
Published online first 16 August 2005
-
Conflict of interest: None declared.