Article Text

Abstract
Cardiovascular complications of cirrhosis include cardiac dysfunction and abnormalities in the central, splanchnic and peripheral circulation, and haemodynamic changes caused by humoral and nervous dysregulation. Cirrhotic cardiomyopathy implies systolic and diastolic dysfunction and electrophysiological abnormalities, an entity that is different from alcoholic heart muscle disease. Being clinically latent, cirrhotic cardiomyopathy can be unmasked by physical or pharmacological strain. Consequently, caution should be exercised in the case of stressful procedures, such as large volume paracentesis without adequate plasma volume expansion, transjugular intrahepatic portosystemic shunt (TIPS) insertion, peritoneovenous shunting and surgery. Cardiac failure is an important cause of mortality after liver transplantation, but improved liver function has also been shown to reverse the cardiac abnormalities. No specific treatment can be recommended, and cardiac failure should be treated as in non-cirrhotic patients with sodium restriction, diuretics, and oxygen therapy when necessary. Special care should be taken with the use of ACE inhibitors and angiotensin antagonists in these patients. The clinical significance of cardiovascular complications and cirrhotic cardiomyopathy is an important topic for future research, and the initiation of new randomised studies of potential treatments for these complications is needed.
Statistics from Altmetric.com
The course in most cirrhotic patients is dominated by complications to portal hypertension, such as bleeding from oesophageal varices and ascites with the development of spontaneous bacterial peritonitis, renal impairment and encephalopathy. Some patients, however, seem to die of causes unrelated to these complications. A closer clinical look shows that a number of these patients display signs of cardiovascular disturbances secondary to vasodilatation, with palmar erythema and reddish skin, raised and bounding pulse, and a low systemic blood pressure indicating a hyperdynamic circulation.1 2 The hyperdynamic syndrome was first described >50 years ago and comprises increased heart rate, cardiac output and plasma volume, and reduced systemic vascular resistance and arterial blood pressure.1–3 The hyperdynamic syndrome is today a well-characterised element in the clinical appearance of the cardiovascular complications of cirrhosis and portal hypertension of various aetiologies.1 2 Experimental and clinical findings of impaired cardiac function have led to the introduction of the new clinical entity, cirrhotic cardiomyopathy, but cardiac dysfunction is not seen in all patients, especially not in those with less advanced disease, and its clinical significance is still under discussion.4 This review will primarily centre on clinical and pathophysiological aspects of the circulatory and cardiac complications of advanced cirrhosis.
THE CIRCULATION IN CIRRHOSIS
An increase in cardiac output can be attributed to an increase in venous return, heart rate and myocardial contractility, all of which are controlled by the autonomic nervous system. Vasodilatation (low systemic vascular resistance), the presence of arteriovenous communications, expanded blood volume and increased sympathetic nervous activity may further raise the cardiac output; most of these pathophysiological mechanisms are active in advanced cirrhosis.1 2 In the early stages, the presence of a hyperdynamic circulation is often not apparent. However, with the progression of the liver disease, there is an overall association between the severity of the cirrhosis and the degree of hyperdynamic circulation. Studies on circulatory changes with posture suggest that the patients are mostly hyperdynamic in the supine position.3 5 6 Blood and plasma volumes are raised in advanced cirrhosis, but the distribution between central and non-central vascular areas is unequal.7 8 Thus, by different techniques it has been established that the central and arterial blood volume—that is, the blood volume in the heart, lungs and central arterial tree—is most often decreased, whereas the non-central blood volume, in particular the splanchnic blood volume, is increased in animals and patients with cirrhosis (see table 1).2 7 9 10 The effective arterial blood volume (ie, the circulatory compartment sensed by baroreceptors) and the central circulation time (ie, central blood volume relative to cardiac output) are substantially reduced and bear a significant relationship to poorer survival in advanced cirrhosis.11
Total vascular compliance as well as arterial compliance (ie, an increase in intravascular volume relative to an increase in transmural blood pressure) are increased in cirrhosis with the degree of decompensation.12 13 The altered static and dynamic characteristics of the wall of large arteries are closely associated with the circulatory and homoeostatic derangement.7 13 14 Arterial compliance depends on the properties of the elastic and smooth muscle of the arterial wall and represents an important coupling between the heart and the arterial system with respect to relocation of intravascular volume.14 The changes in arterial mechanics are reversible at least in part.13 An element in the elevated arterial compliance in advanced cirrhosis is the reduced arterial blood volume and blood pressure.7 Arterial compliance expresses the stroke volume relative to the pulse pressure and is directly related to the severity of cirrhosis.13 14 However, in contrast to the systemic vascular resistance, arterial compliance may be determined independently of flow and pressure by pulsewave velocity.14 In addition, the arterial compliance in cirrhosis seems to be affected by vasoactive forces as it correlates directly with the vasodilator calcitonin gene-related peptide (CGRP) and inversely with catecholamines.13 The arteriolar tone adjusts the level of the blood pressure and may thereby influences large artery compliance. Recent data suggest that the hyperdynamic circulation is mainly caused by circulatory alterations in the splanchnic area.15 Thus, arteriolar vasodilatation would be a more localised event, whereas the elevation in arterial compliance may be more systemic.7 Arterial compliance may therefore be an integral variable for vascular responsiveness, together with the systemic vascular resistance. Arterial compliance is easy to determine and elevated in advanced cirrhosis. Besides a relationship to age, body size, sex and the level of arterial blood pressure, arterial compliance is directly related to the severity of cirrhosis, the hyperdynamic circulatory derangement and abnormal volume distribution. Its role in clinical hepatology, however, remains to be established.
The pulmonary vascular resistance is often decreased in cirrhosis, except in the 2–4% of the patients with portopulmonary hypertension.16 Some patients exhibit characteristic vascular abnormalities with arteriovenous shunts and intrapulmonary dilatations.1 16 Ventilatory lung function and diffusion are impaired in the majority of the patients, and the combination of vascular abnormalities, reduced transfer factor and low arterial oxygen saturation has been termed the hepatopulmonary syndrome.16 In patients with cirrhosis, the reduced transfer factor correlates with the low pulmonary blood volume, which suggests that central underfilling also plays a role in the impairment of pulmonary function.17
Pathophysiology of splanchnic arteriolar vasodilatation
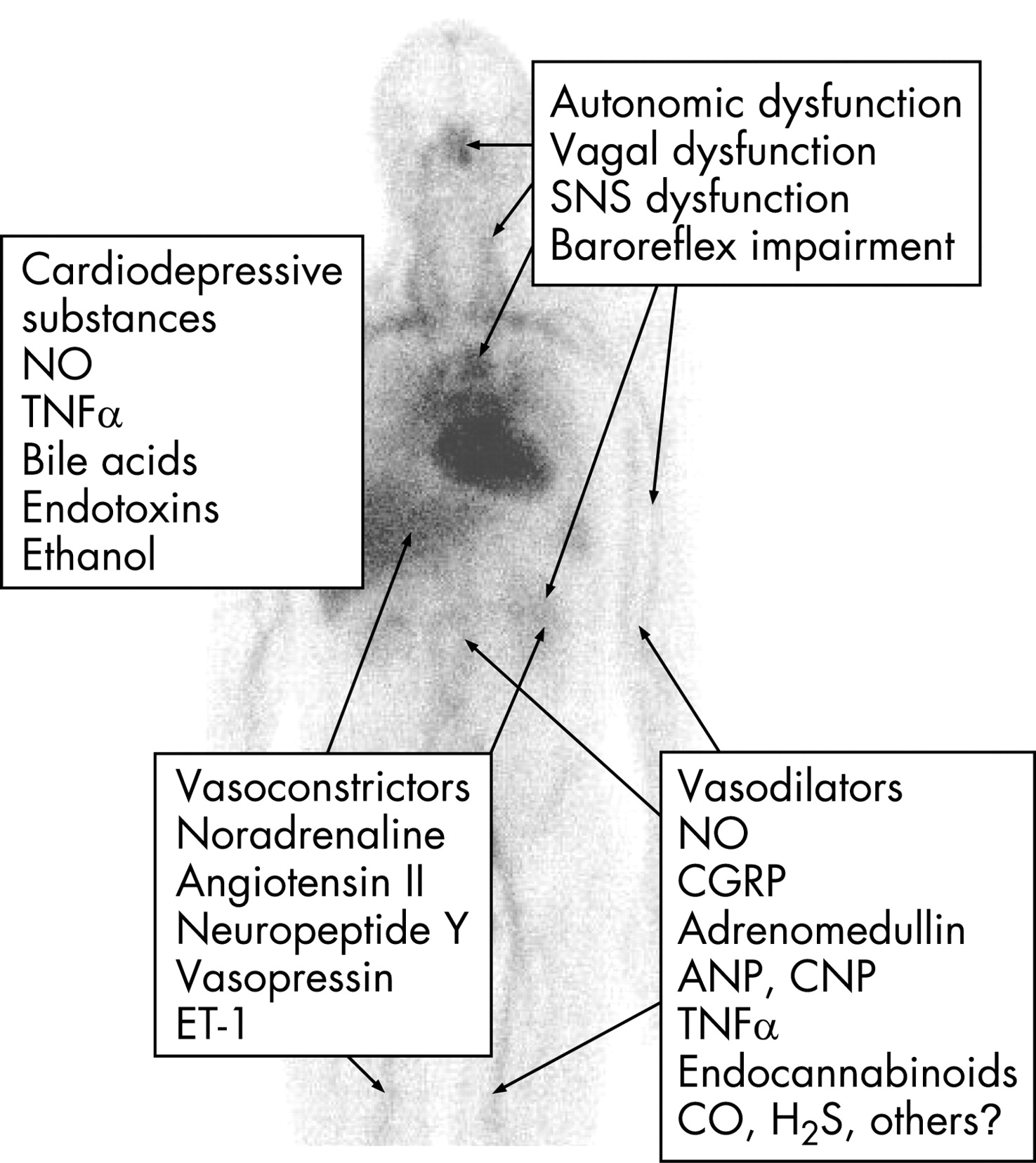
Arteriolar vasodilatation in cirrhosis and portal hypertension may be brought about by a combination of overproduction of circulating vasodilators, vasodilators of intestinal or systemic origin, vasodilators that escape degradation in the diseased liver or bypass the liver through portosystemic collaterals, reduced resistance to vasoconstrictors and increased sensitivity to vasodilators.1 2 According to “the arterial vasodilation hypothesis”, splanchnic arteriolar vasodilation leads to reduction of the systemic vascular resistance, central arterial underfilling with effective hypovolaemia, activation of vasoconstrictor systems, such as the sympathetic nervous system (SNS), the renin–angiotensin–aldosterone system (RAAS), vasopressin, endothelins (ETs) and neuropeptide Y, and hence development of a hyperkinetic circulatory state.1 8 18 Thus, most of the haemodynamic changes summarised in table 1 and figure 1 can be explained by this theory. The predominantly splanchnic vasodilation in cirrhosis precedes the increase in cardiac output and heart rate, and it has recently been shown experimentally that mild increases in portal pressure upregulate nitric oxide synthase (eNOS).19 With the progression of the disease, the splanchnic vasodilatation becomes more pronounced and the hyperdynamic circulation may no longer be sufficient to correct the effective hypovolaemia.20 21 The splanchnic circulation is less sensitive to the effects of angiotensin II, noradrenaline and vasopressin because of the surplus of vasodilators which may play a role in the development of the vascular hyporesponsiveness to vasoconstrictors.22 The arterial blood pressure is mainly maintained by vasoconstriction in the renal, cerebral and hepatic vascular beds where there seems to be a diminished release of nitric oxide (NO) from endothelial cells.15 23

To explain the vasodilatation in the systemic circulation, recent research has focused especially on substances such as NO, CGRP and adrenomedullin, but natriuretic peptides, interleukins, hydrogen sulphide, ETs and endocannabinoids have also been implicated (table 2).1 Blockade of NO formation in animal models and cirrhotic patients significantly increases arterial blood pressure and decreases plasma volume, sodium retention and forearm blood flow.24 25 Taken together, there is a growing body of evidence that systemic NO production is increased and precedes the development of the hyperdynamic circulation in cirrhosis, thereby playing a major role in the arteriolar and splanchnic vasodilation and vascular hyporeactivity.15 In addition, vascular endothelial growth factor (VEGF) seems to stimulate angiogenesis and the development of portosystemic collaterals, and blockade of the VEGF receptor-2 has been shown experimentally to inhibit this process and revert portal hypertension and the hyperdynamic circulation.19 26 In addition, recent studies have suggested that the haem oxygenase–carbon monoxide pathway mediates hyporeactivity to phenylephrine in splanchnic vessels.27 CGRP and adrenomedullin are powerful vasodilating peptides, which are both elevated in cirrhosis, especially in those patients with ascites and the hepatorenal syndrome correlating with markers of central hypovolaemia.1 Hydrogen sulphide is a gaseous transmitter with potent vasodilating properties, which has recently been implicated in vascular abnormalities in cirrhosis.28 New experimental data suggest that defective rho-kinase signalling may also contribute to the hypocontractility in cirrhosis.29 Thus, the excess of vasodilators combined with an inadequate haemodynamic response to vasoconstrictors may explain the vasodilatatory state and vascular hyporeactivity in cirrhosis combined with a hyperdynamic circulation, but the pathophysiological mechanisms behind the development of the hyperdynamic circulation in cirrhosis may be multifarious, as listed in table 3.
The hepatic circulation
From a haemodynamic point of view, the hepatic vascular resistance and portal inflow determine the level of portal pressure. Factors that determine the hepatic vascular resistance include both structural and dynamic components. Among the structural components are histological characteristics such as steatosis, fibrosis and regeneration nodules. Dynamic structures include cells with contractile properties such as hepatic stellate cells, myofibroblasts and smooth muscle cells.30 Portal venous inflow is mainly determined by the degree of splanchnic vasodilation. In healthy subjects, the hepatic blood flow equals the splanchnic blood flow, but patients with portal hypertension have a substantial portosystemic collateral circulation, and an increased mesenteric inflow of up to several litres per minute has been reported (table 1). Thus, a large part of the increased cardiac output is returned through portosystemic collaterals. The azygous blood flow is especially important, as the azygous vein drains oesophageal varices and an increase in azygous flow is associated with an increased risk of variceal bleeding.11 β-Blockers, nitrates, octreotide, terlipressin, etc. can reduce the increased splanchnic blood flow pharmacologically, and infusion of these drugs may in some patients partially reverse the hyperkinetic mesenteric circulation. As outlined above, there seems to be a defective sinusoidal eNOS-derived production of NO.15 In addition, recent investigations of endogenous vasoactive substances have focused especially on ET-1, angiotensin II, catecholamines and leukotrienes in the increased hepatic–sinusoidal resistance.1 30 The haemodynamic imbalance with a predominant sinusoidal constriction may contribute significantly to the development of portal hypertension and be an important target for treatment.
Volume distribution and circulatory dysfunction
Imbalance between vasodilating and vasoconstricting forces in cirrhosis contributes to an abnormal distribution of volume, vascular resistance and flow. Although the cardiac output is increased, thereby reflecting substantial vasodilatation, it covers hyperperfused, normoperfused and hypoperfused vascular beds. Thus, in the kidney, vasoconstriction prevails and plays a pivotal role along with the development of hepatic decompensation. Liver dysfunction, central hypovolaemia, arterial hypotension and neurohumoral activation of particularly the RAAS and SNS with renal vasoconstriction is of major importance.1 20 The increased plasma volume in cirrhosis should therefore be considered secondary to the activation of neurohumoral mechanisms consequent on mainly splanchnic vasodilatation, low arterial blood pressure and reduced central and arterial blood volume.
Central hypovolaemia and arterial hypotension may be ameliorated by infusion of plasma expanders. During volume expansion, most cirrhotic patients respond with a further reduction in systemic vascular resistance rather than an increase in arterial blood pressure.7 9 The infusion of hyperosmotic solutions or albumin in cirrhosis results initially in a shift of fluid from the interstitial space into the plasma volume, with expansion of the latter.7 9 Irrespective of severity, volume expansion produces a rise in stroke volume and cardiac output. In early cirrhosis there is a proportional expansion of the central and non-central parts of the blood volume, whereas in late cirrhosis, expansion is mainly confined to the non-central part, with a proportionally smaller increase in cardiac output, probably because of cardiac dysfunction and abnormal vascular compliance.9 31 Similar effects are seen after infusion of a plasma protein solution, whereas infusion of packed red blood cells may be less efficient possibly because of a difference in the trapping of NO and shear stress.1
When therapeutic paracentesis is done in decompensated cirrhosis without administration of plasma expanders, about 75% of patients will develop what is termed paracentesis-induced circulatory dysfunction.32 This condition is characterised by a pronounced activation of the RAAS and SNS, which reflects central hypovolaemia. It is mainly caused by a paracentesis-induced splanchnic arteriolar vasodilatation and brings about a further reduction in the systemic vascular resistance.33 Intravenous infusion of albumin has been shown to prevent complications caused by circulatory dysfunction and may prevent development of renal failure and rapid occurrence of ascites, and prolong survival.32 Recent studies have shown, however, that administration of vasoconstrictors such as terlipressin or noradrenaline may be effective alone or especially in combination with albumin.34 35 Paracentesis-induced circulatory dysfunction is thus an example of a cirrhotic condition where complications attributable to a potentially reduced effective blood volume can be prevented by a specific volume expansion.
The deterioration of the liver function is followed by a decreased renal blood flow and glomerular filtration rate, and increased sodium and water reabsorption, and may progress into the hepatorenal syndrome, a functional and reversible renal impairment in severely ill patients (table 1).20 However, glomerular hyperfiltration has been described in some patients with preascitic cirrhosis.36 Recently, a new concept has been put forward in the pathophysiological explanation of renal dysfunction as a circulatory dysfunction characterised by insufficient cardiac output leading to effective hypovolaemia.20 21 This concept is supported by data from a longitudinal study in non-azotaemic cirrhotic patients suggesting that circulatory dysfunction with a decrease in cardiac output combined with splanchnic arterial vasodilatation and activation of the RAAS contribute to renal dysfunction and the hepatorenal syndrome.20 37 Angiotensin II mainly acts on the efferent arteriole, and a low dose of an ACE inhibitor may induce a significant reduction in glomerular filtration and a further reduction in sodium excretion, even in the absence of a change in arterial blood pressure. This suggests that the integrity of the RAAS is important for the maintenance of renal function in cirrhotic patients and that RAAS overactivity does not solely contribute to the adverse renal vasoconstriction. Treatment of the hepatorenal syndrome is directed towards improving liver function by liver transplantation, arterial hypotension and central hypovolaemia, and reducing renal vasoconstriction, for instance with the combined use of splanchnic vasoconstrictors such as terlipressin and plasma expanders such as human albumin.20
The circulation of the extremities
The cutaneous and muscular circulations may be increased in patients with cirrhosis.1 Palmar erythema, spider naevi and potatory face were early recognised as clinical signs of cutaneous hyperperfusion. These types of circulatory abnormalities illustrate capillary hyperperfusion and the presence of arteriovenous fistulae. Muscular circulation is reported to be increased, normal and reduced in patients with cirrhosis.38 39 Evaluation of brachial and femoral artery blood flow by Doppler techniques has failed to disclose a clear hyperdynamic perfusion of the limbs.38 39 Recently, however, it has been shown that blockade of NOS causes peripheral vasoconstriction in the forearm in cirrhosis and that this system contributes in the regulation of the peripheral vascular tone and to the hyperdynamic state.25 Estimates of skin blood flow by nuclear medicine techniques have shown normal capillary skin blood flow in cirrhotic patients.40
The techniques used are hampered by various caveats relating to the methods in use and experimental circumstances. Venous occlusion plethysmography with forearm and leg measurements may give a combination of cutaneous and muscular blood flow, but this method has also given identical baseline values in patients and controls.41 We still have only a faint impression of the haemodynamics of the peripheral circulation in cirrhosis, and the cutaneous and muscular circulations in cirrhosis are important topics for further research. At present it can be concluded that the increased cardiac output in patients with cirrhosis covers systemic vascular beds with various degrees of perfusion, owing to an imbalanced state of vasoconstriction and vasodilatation. The exact distribution of the increased cardiac output to the different organs, tissues and types of vessels remains to be clarified.
ABNORMALITIES IN THE REGULATION OF THE CIRCULATION
Autonomic dysfunction
Cirrhosis is often associated with autonomic neuropathy which has become evident from studies of haemodynamic responses to standard cardiovascular reflex tests, such as heart rate variability and isometric exercise.3 5 42 Most studies on these issues have found a high prevalence of autonomic dysfunction in cirrhosis with associations with liver dysfunction and survival.43 44 The autonomic dysfunction may be temporary, arises as a consequence of liver dysfunction and seems reversible after liver transplantation.45 Most studies have focused on defects in the SNS, but the importance of vagal impairment for sodium and fluid retention has been shown.3 42 43 Sympathetic responses to exercise are clearly impaired.46 47 Similarly, blood pressure responses to orthostasis are impaired, probably because of a blunted baroreflex function in advanced cirrhosis.5 48 Abnormal cardiovascular responses to vasoconstrictors have been reported in patients with cirrhosis,1 and there is experimental evidence that haem oxygenase mediates hyporeactivity to phenylephrine in the mesenteric vessels of cirrhotic rats with ascites.27 Administration of captopril partly corrects the parasympathetic dysfunction in cirrhosis, which indicates that the vagal component is to a certain extent caused by neuromodulation with angiotensin II.43 Involvement of the RAAS is also supported by data that show normalisation of cardiac responses to postural changes after administration of canrenone, an aldosterone antagonist, to compensated cirrhotic patients.48 Interestingly, the vasoconstrictor hyporeactivity seems to be reversible by such antioxidants as vitamin C, which indicates that oxidative stress plays a role in vascular hyporeactivity and that antioxidant therapy could possibly have a role in these complications in cirrhosis.49
The pathophysiological basis underlying the autonomic dysfunction in cirrhosis is unknown, but relationships to the severity of the liver disease, mortality and reversibility after liver transplantation point to hepatic metabolism and increased NO production, and reduced vasoconstrictor sensitivity with postreceptor defects. This provides some explanation for the vascular hyporeactivity in cirrhosis (fig 2).
Arterial blood pressure and baroreceptor function in cirrhosis
The level of the arterial blood pressure, which depends on the cardiac output and the systemic vascular resistance, is kept low normal in cirrhosis as a circulatory compromise between the vasodilatating and counter-regulatory vasoconstricting forces affecting both vascular resistance and arterial compliance. There is a relationship between the degree of arterial hypotension in cirrhosis and the severity of disease, signs of decompensation, and survival.1 11 SNS, RAAS, vasopressin and ET-1 are all important vasoconstrictors involved in the maintenance of the arterial blood pressure in cirrhosis.1 50 The impact of potent vasodilators has been mentioned above. NOS blockade causes higher arterial blood pressure in cirrhotic rats and reduces forearm blood flow in cirrhotic patients.25 Inhibition of the endocannabinoid CB1 receptor raises arterial blood pressure and cardiac contractility in experimental cirrhosis, and anandamide increases the splanchnic vessel diameter, flow and cardiac output and may thus contribute to the hyperkinetic state and arterial hypotension in cirrhosis.51–53 The arterial blood pressure possesses a circadian variation. In cirrhosis, the arterial blood pressures are reduced during the day, whereas at night the values are normal, which indicates an abnormal blood pressure regulation.54 A resetting of the baroreceptors is still discussed in human conditions in relation to wall tension of the fibroelastic tissues in the vessels and stretch-induced activation of the sodium–potassium channels.8 Whereas the baroreflex sensitivity (BRS) may be normal in early cirrhosis,55 there is substantial evidence that BRS is impaired in patients with advanced disease.56 57 Recently, we have described relationships of the reduced BRS to determinants of the central circulation and the RAAS. Together with a flat blood pressure/heart rate slope as found during 24 h ambulatory blood pressure monitoring, this indicates that low BRS contributes to the dysregulation of the arterial blood pressure, although the precise mechanism is unknown.54 57
CARDIAC DYSFUNCTION IN CIRRHOSIS
The expanded blood volume in advanced cirrhosis contributes to a persistent increase in cardiac output, which may overload the heart.58 In other circumstances, increased cardiac output and augmented cardiac work would cause cardiac failure but, because of the decreased afterload, as reflected by reduced systemic vascular resistance and increased arterial compliance, left ventricular failure may be latent in cirrhosis.4 13 59 Cardiac failure may become manifest under strain or treatment with vasoconstrictors. This type of cardiac dysfunction has been termed “cirrhotic cardiomyopathy” and was for years erroneously attributed to alcoholic heart muscle disease. At the 2005 World Congress of Gastroenterology at Montreal, a working party of experts in the field was set up to work out a classification system for cirrhotic cardiomyopathy. Essentials in the definition are a chronic cardiac dysfunction in cirrhotic patients, characterised by blunted contractile responsiveness to stress, and/or altered diastolic relaxation with electrophysiological abnormalities in the absence of other known cardiac disease (table 4), and a consensus working group is developing a specific definition to be published in 2008. Elements in cirrhotic cardiomyopathy include impaired cardiac contractility with a systolic dysfunction, diastolic dysfunction and electromechanical abnormalities with a prolonged Q–T interval.4 59 Various electrophysiological mechanisms for the conductance abnormalities and impaired cardiac contractility have been suggested and include changes in the cardiomyocyte plasma membrane with an increased cholesterol/phospholipid ratio, attenuated function of the β-adrenergic pathway and greater activity of inhibitory systems.4 Other studies have focused on negative inotropic effects of NO, nitration of cardiac proteins, CO, endogenous cannabinoids, bile acids, endotoxins and other systems.59 60 Cannabinoids are endogenous ligands including anandamide that binds to cannabinoid receptors CB1 and CB2.4 51 The production may increase in response to stress such as tachycardia and overload.61 Experimental studies have shown a negative inotropic effect of anadamide in cirrhotic rats, which suggests that this system is involved in cirrhotic cardiomyopathy.4 62 The haem oxygenase–CO pathway has also been shown to play a role in the pathogenesis of abnormal cardiac contractility in cirrhotic cardiomyopathy.4 27
Systolic dysfunction
In cirrhotic cardiomyopathy, the left ventricular end-diastolic pressure increases after exercise, but the expected increases in cardiac stroke index and left ventricular ejection fraction (LVEF) are absent or subnormal, which indicates an inadequate response of the ventricular reserve to a rise in ventricular filling pressure.63 A vasoconstrictor-induced increase of 30% in the left ventricular afterload results in an approximate doubling in pulmonary capillary wedge pressure, with no change in cardiac output.31 Recently, we have shown by myocardial perfusion imaging that infusion of terlipressin suppresses myocardial function, whereas the myocardial perfusion is left unaffected.64 This response may be useful in diagnosing cirrhotic cardiomyopathy. A similar pattern is seen after insertion of a transjugular intrahepatic portosystemic shunt (TIPS), but the raised cardiac pressures after TIPS tend to normalise with time.65 66 Some of these patients (12%) may develop manifest cardiac failure in association with the TIPS insertion.67 Similar effects are seen after infusion of plasma expanders. Infusion of a plasma protein solution, however, increases cardiac output, as well as right atrial pressure, pulmonary arterial pressure and pulmonary capillary wedge pressure, whereas infusion of packed red blood cells may not produce a change in these variables.1
The LVEF reflects systolic function, even though it is very much influenced by preload and afterload. It has been reported to be normal at rest in some studies and reduced in one study of a subgroup of patients with ascites.31 63 68 After exercise, LVEF increases less in cirrhotic patients than in controls (fig 3).59 63 69 The reduced functional capacity may be attributed to a combination of blunted heart rate response to exercise, reduced myocardial reserve and profound skeletal muscle wasting with impaired oxygen extraction.46 47 In patients with advanced cirrhosis and severe vasodilatation, activation of the RAAS, impaired renal function and a reduced systolic function (a decrease in cardiac output) appear to be major determinants for the development of the hepatorenal syndrome.37 Spontaneous bacterial peritonitis is a well-known risk factor for the development of the hepatorenal syndrome, and after resolution of the infection suppression of systolic function appears to be more pronounced in patients who develop renal failure. Maintenance of cardiac contractility thus appears to be an important factor in the prevention of renal failure.70

Diastolic dysfunction
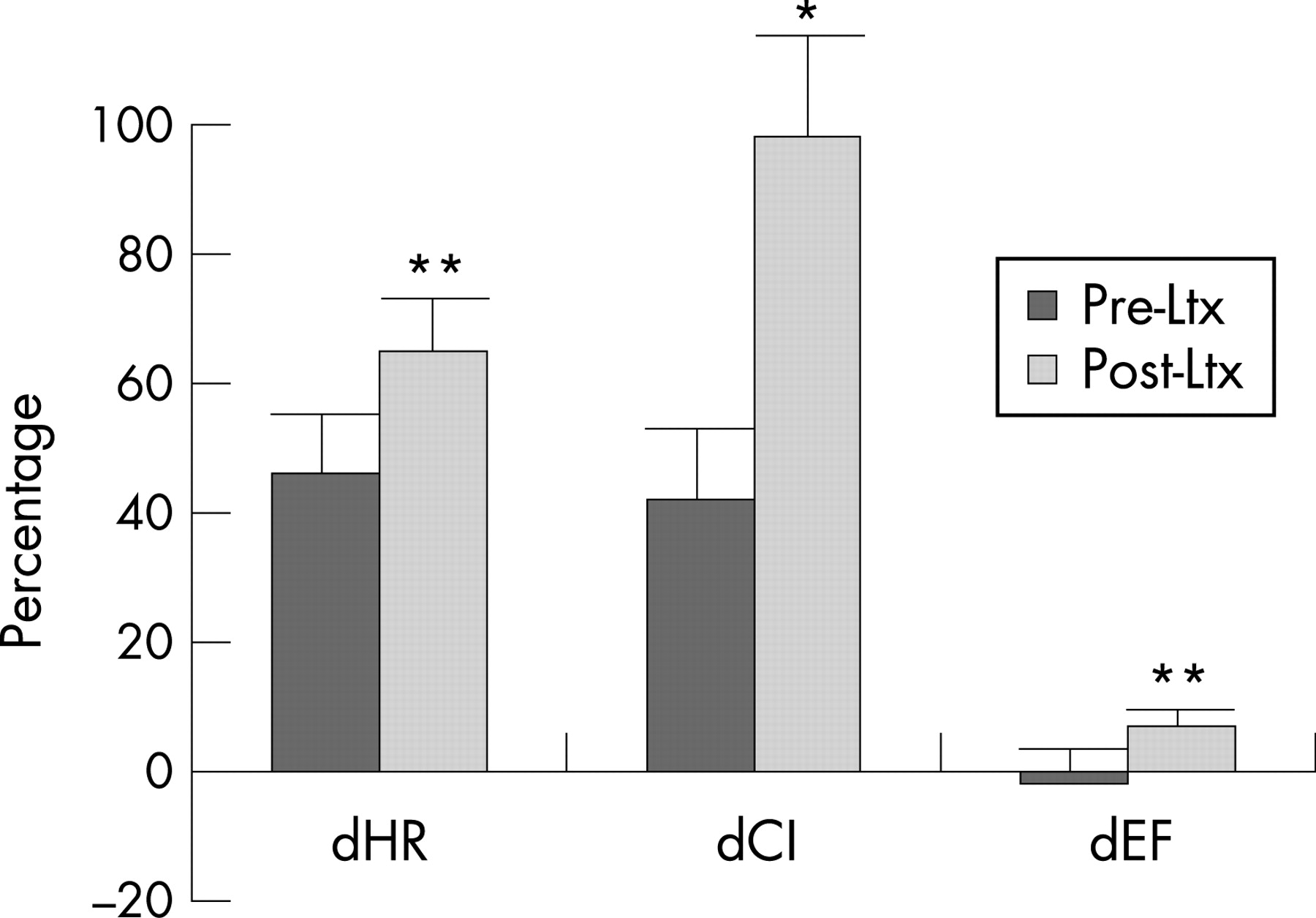
Many patients with cirrhosis exhibit various degrees of diastolic dysfunction, which implies changes in myocardial properties that affect left ventricular filling. Diastolic dysfunction may progress to systolic dysfunction, although this has not been directly shown in cirrhotic patients.31 71 The pathological basis of the increased stiffness of the left ventricle seems to be cardiac hypertrophy, patchy fibrosis and subendothelial oedema.4 31 69 Determinants of a diastolic dysfunction on a Doppler echocardiogram are decreased E/A ratio (the ratio of early to late (atrial) phases of ventricular filling) and delayed early diastolic transmitral filling with prolonged deceleration and isovolumetric relaxation times (table 4).31 68 72 In a number of studies, A wave and E wave velocities and deceleration times are much increased and the E/A ratio is decreased in cirrhotic patients, especially in those with ascites.68 72 Recent studies of ventricular diastolic filling in cirrhosis support the presence of a subclinical myocardial disease with diastolic dysfunction, which, in ascitic patients, improves after paracentesis and can be aggravated after TIPS.65 68 72 In these decompensated patients, paracentesis seems to ameliorate diastolic, but not systolic, function.68 Patients with TIPS with an E/A ratio <1 seem to have a poorer survival rate than patients without signs of diastolic dysfunction.73 Liver transplantation has recently been shown to reverse cardiac changes, including diastolic dysfunction (fig 4).69 It has been proposed that diastolic dysfunction precedes systolic dysfunction in early heart disease and that anti-aldosterone treatment improves cardiac function. Pozzi et al recently demonstrated that anti-aldosterone treatment with K-Canrenoate in cirrhosis ameliorated cardiac structure by reducing left ventricular wall thickness and volume, but had almost no effects on systolic and diastolic functions.74 It is also possible that anti-aldosterone treatment may have beneficial effects on catecholamine-induced cardiac fibrosis, as described in heart failure.75

The clinical significance of diastolic dysfunction and its importance in cirrhotic cardiomyopathy has been questioned, as overt cardiac failure is not a prominent feature of cirrhosis. However, there are several reports of unexpected death from heart failure following liver transplantation, surgical portocaval shunts and TIPS.67 76 These procedures involve a rapid increase in cardiac preload. In a less compliant heart, the diastolic dysfunction could be enough to cause pulmonary oedema and heart failure. This is consistent with the findings of Huonker et al,65 who reported an increase in pulmonary artery pressure, preload and diastolic dysfunction after TIPS. In patients with the hepatopulmonary syndrome and in children with chronic hepatitis, an isolated right ventricular diastolic dysfunction has been described and may play a role in the right cardiac function and course of these patients.77 Thus, both left and right diastolic dysfunction could account for part of the cardiac dysfunction in cirrhotic cardiomyopathy.
Electromechanical abnormalities
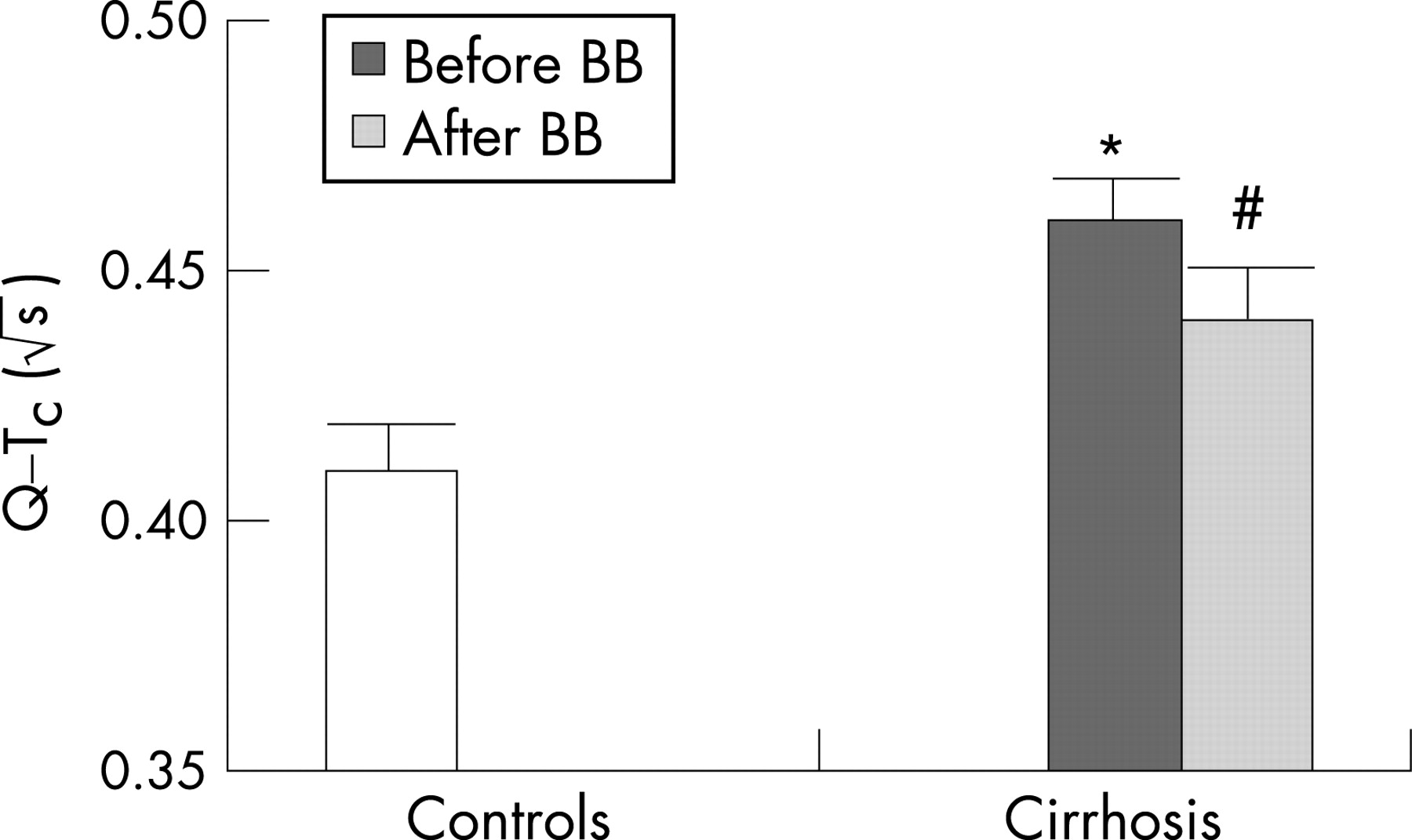
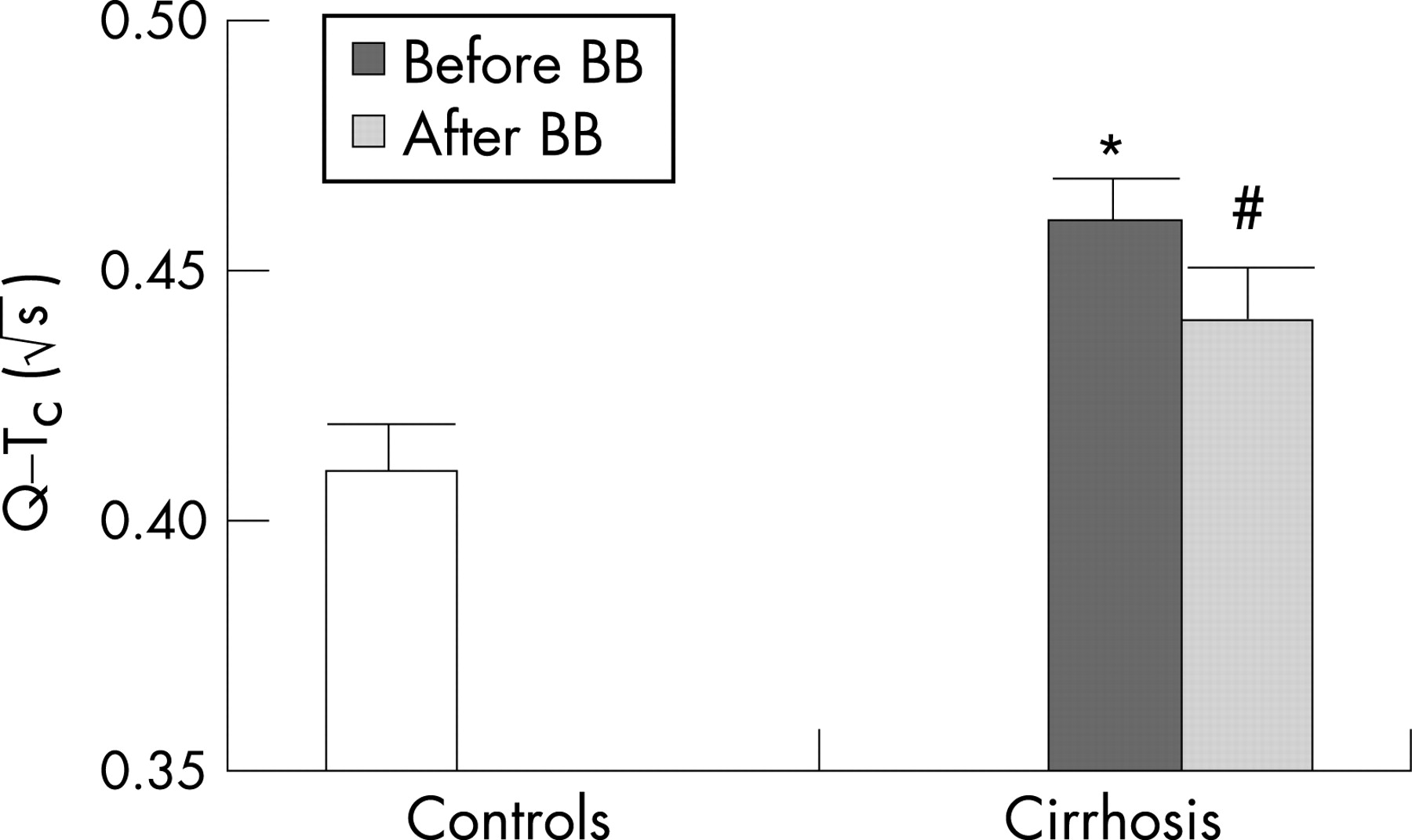
There is a large body of evidence for electrophysiological abnormalities in cirrhosis primarily comprising prolonged repolarisation time and increased dispersion of the electromechanical time interval.78 79 The sympathetic nervous activity influences the heart rate and electromechanical coupling by several mechanisms: noradrenaline binding to β-receptors, receptor-mediated G protein interaction and, consequently, stimulation of adenylcyclase, activation of cAMP-dependent phosphokinase A and channel phosphorylation. Several receptor and postreceptor defects have been described in cirrhosis with reduced β-receptor density and sensitivity, and altered G protein and calcium channel functions.4 80 All these defects may explain both impaired chronotropic responses and electromechanical uncoupling. The coupling between cardiac contractions and the arterial system is of major importance for the amount of work performed by the left ventricular myocardium, and thereby for the strain on the heart.14 46 In addition, Ward et al have shown a decrease in K+ currents in ventricular cardiomyocytes from cirrhotic rats, which prolongs the Q–T interval.81 The prolonged repolarisation time is reflected by a prolonged Q–T interval in a substantial fraction of the patients with cirrhosis, which could lead to ventricular arrhythmias and sudden cardiac death, but the evidence from clinical studies is sparse.4 59 In cirrhotic patients, the prolonged Q–T interval is significantly related to the severity of the liver disease, portal hypertension, portosystemic shunts, elevated brain-type natriuretic peptide (BNP) and pro-BNP, elevated plasma noradrenaline and reduced survival.79 82 83 The prolongation of the Q–T interval is partly reversible after liver transplantation and β-blocker treatment (fig 5).45 82 The prolonged Q–T interval in cirrhosis should be considered an element in the cirrhotic cardiomyopathy and may be of potential use in identifying patients at risk.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CONCLUDING COMMENTS
Cardiovascular complications in cirrhosis may arise on the basis of combined humoral, nervous and haemodynamic changes. Cirrhotic cardiomyopathy suggests a systolic and diastolic dysfunction and electrophysiological abnormalities. It is different from alcoholic heart muscle disease and appears to be unmasked by procedures that stress the heart, such as pharmacological vasoconstriction, exercise, and by insertion of TIPS (Box 1).59 Potential diagnostic tools primarily include echocardiography and ECG (table 5). The cardiovascular complications in cirrhosis and cirrhotic cardiomyopathy may be part of a multiorgan syndrome that affects the patients’ prognosis.1 2 No specific treatment can be recommended, and is largely empiric and supportive. Caution should be exercised with respect to stressful procedures, such as large volume paracentesis without adequate plasma volume expansion, TIPS insertion, peritoneovenous shunting and surgery.4 Cardiac failure is an important cause of mortality after liver transplantation. On the other hand, liver transplantation has been shown to reverse systolic and diastolic dysfunction and the prolonged Q–T interval.69 Thus, although the post-transplant pathophysiological mechanisms are complex, liver transplantation appears to be an effective treatment of the cardiovascular complications of cirrhosis.
Box 1: Key points of cirrhotic cardiomyopathy
Present in the face of a hyperkinetic circulation with a combined systolic and diastolic dysfunction together with electrophysiological abnormalities.
Different from alcoholic heart muscle disease
Systolic dysfunction demasked by physical or pharmacological stress
Diastolic dysfunction detected by echocardiographic measurement of the E/A ratio
Q–T interval prolongation assessed on the ECG and adjusted adequately
Treatment is non-specific and directed towards the left ventricular heart failure
Improvement of left ventricular contractility with ACE inhibitors should be done with care, as this may provoke severe arterial hypotension. β-Blockers have been shown to reduce acutely the prolonged Q–T interval and may, in addition to the cardioprotective effects, be of benefit.79 82 However, effects on morbidity and mortality remain to be shown in longitudinal studies.
Future studies should be directed towards a delineation of the clinical importance of cardiovascular complications and cirrhotic cardiomyopathy, and randomised to examine benefits of the treatments outlined above.
REFERENCES
Footnotes
Competing interests: None.