Article Text

Abstract
Refractory coeliac disease (RCD) is defined by persistent or recurrent malabsorptive symptoms and villous atrophy despite strict adherence to a gluten-free diet (GFD) for at least 6–12 months in the absence of other causes of non-responsive treated coeliac disease and overt malignancy. Symptoms are often severe and require additional therapeutic intervention besides a GFD. RCD can be classified as type 1 (normal intraepithelial lymphocyte phenotype), or type 2 (defined by the presence of abnormal (clonal) intraepithelial lymphocyte phenotype). Patients with RCD may never have responded to a GFD or may have relapsed despite adherence and initial response to the GFD. RCD type 1 usually improves after treatment with a combination of aggressive nutritional support, adherence to a GFD, and alternative pharmacological therapies. By contrast, clinical response to alternative therapies in RCD type 2 is less certain and the prognosis is poor. Severe complications such as ulcerative jejunitis and enteropathy-associated T cell lymphoma may occur in a subgroup of patients with RCD. The aims of this article are to (1) review recent advances in the diagnosis and management of patients with RCD, and (2) describe current and novel methods for classification of patients with RCD into categories that are useful to predict outcome and direct treatment.
- Lymphoma
- sprue
- clone
- serology
- intraepithelial lymphocytes
- ASCT
- autologous hematopoietic stem cell transplantation
- CD
- coeliac disease
- EATL
- enteropathy-associated T cell lymphoma
- EMA
- endomysial antibodies
- GFD
- gluten-free diet
- HLA
- human leucocyte antigens
- MRI
- MRI
- PET
- positron emission tomography
- RCD
- refractory coeliac disease
- tTGA
- tissue transglutaminase antibodies
Statistics from Altmetric.com
- ASCT
- autologous hematopoietic stem cell transplantation
- CD
- coeliac disease
- EATL
- enteropathy-associated T cell lymphoma
- EMA
- endomysial antibodies
- GFD
- gluten-free diet
- HLA
- human leucocyte antigens
- MRI
- MRI
- PET
- positron emission tomography
- RCD
- refractory coeliac disease
- tTGA
- tissue transglutaminase antibodies
Background
Coeliac disease is an immune-mediated disorder affecting genetically predisposed subjects, and is caused by the ingestion of gluten present in cereals such as wheat, barley and rye.1 The disease affects around 1% of the general population in developed and developing countries, with increasing prevalence over time reported in the United States and Europe.2–4 A lifelong gluten-free diet (GFD) is the only effective treatment to alleviate the symptoms, normalise antibodies and the intestinal mucosa in patients with coeliac disease.5
Clinical response is observed in most patients with coeliac disease after only a few weeks on a GFD.6 However, complete clinical response and mucosal recovery does not occur in all patients with treated coeliac disease.7 Indeed, a subgroup of patients with coeliac disease may have persistent or recurrent symptoms (eg, diarrhoea, abdominal pain and weight loss), inflammation of the intestine, and villous atrophy despite strict adherence to a GFD.8 9 Symptoms are often severe and require additional therapeutic intervention besides GFD.5 8 RCD is defined by persistent or recurrent malabsorptive symptoms and villous atrophy despite strict adherence to a GFD for at least 6–12 months in the absence of other causes of non-responsive treated coeliac disease and overt malignancy.10–12 The aims of this article are to (1) review recent advances in the diagnosis and management of patients with RCD, and (2) describe current and novel methods for classification of patients with RCD into categories that are useful to predict outcome and direct treatment.
Material and methods (review criteria)
PubMed was searched in July 2009 for full articles published in English-language journals with the following keywords alone or in combination: ‘coeliac disease’, ‘aberrant lymphocytes’, ‘refractory sprue’, ‘clonality’, ‘refractory coeliac disease’, ‘unresponsive coeliac disease’, ‘gluten-free diet’, ‘intractable diarrhoea’, ‘malignancy’, and ‘lymphoma’. In this literature search, several points became obvious: (1) the prevalence of refractory coeliac disease (RCD) in the community is unknown; (2) a precise definition of RCD is lacking and the diagnostic criteria for RCD vary from centre to centre; (3) the mechanisms underlying RCD are poorly understood; (4) the natural history, clinical evolution, and prognostic factors in RCD require further study; (5) prospective multicentre clinical trials to test novel therapies are needed; (6) most recommendations for evaluation and treatment are based on expert opinion, not evidence-based reasoning; (7) there are well-described, relatively large case series from referral centres; (8) there are emerging data that might improve current classification and clinical staging of RCD; and (9) novel therapies for patients with RCD type 2 are needed to effectively control symptoms and reduce complications particularly progression to lymphoma. Citations were chosen on the basis of their relevance to the text.
Epidemiology
The real prevalence of RCD is unknown but is probably rare. Evidence of the rarity of RCD is the low number of cases reported in the literature, most often from major coeliac disease referral centres.13–18 However, RCD may be the cause underlying persistent or recurrent symptoms in treated coeliac disease in just 10–18% of the patients evaluated in referral centres.10 11
Estimates of the occurrence of RCD in non-referral, population-based cohorts are very scarce. RCD was diagnosed in only five (0.7%) of 713 patients with coeliac disease from the Derby cohort (United Kingdom) from 1978 to 2005.19 From 204 biopsy-confirmed coeliac disease residents of Olmsted County (Minnesota, United States) identified from 1950 to 2006, only three (1.47%; 95% CI, 0.3% to 4.2%) had a subsequent diagnosis of RCD type 1 (n=2) or type 2 (n=1). The incidence per 100 000 person-years was 0.06 (95% CI, 0.0 to 0.12) adjusted for age and gender to the 2000 US white population (A.R-T, unpublished data 2009) Thus, RCD appears to be an uncommon condition but with a poor outcome.1
RCD affects two to three times as many women than men,13 15 17 consistent with the predominance of diagnosed coeliac disease in adult women.1 The predominance of the disease in women diminishes somewhat in those patients with both RCD and enteropathy-associated T cell lymphoma (EATL).13 17 A diagnosis of RCD is exceptional before the age of 30 years and most cases are diagnosed around the age of 50 years or thereafter.15 17
Clinical manifestations
Persistent diarrhoea, abdominal pain, and involuntary loss of weight are the most common symptoms in RCD.20 Multiple vitamin deficiencies, anaemia, fatigue, malaise, thromboembolic events, and coexisting autoimmune disorders are also frequent. 8 14 20 The majority of patients with RCD are diagnosed because of the development of new symptoms or recurrence of diarrhoea after initial clinical response to GFD for years (‘secondary’ RCD).15 17 However, a subgroup of patients is diagnosed because of the necessity of early intervention to control their symptoms due to lack of response after 6–12 months of GFD (‘primary’ RCD).15 17
Laboratory findings
Low haemoglobin and hypoalbuminaemia are frequent findings and may indicate a poor prognosis.13 17 Chronic elevated levels of transaminases were detected in one half of patients in one series.15 High stool output (median of 1 l/day) often associated with significant steatorrhoea is common.17 Human leucocyte antigen (HLA)-DQ2 is present in up to 98% of the cases, with HLA-DQ8 present in almost all others.15 17
While coeliac disease-specific serology (either endomysial or tissue transglutaminase antibodies) is usually positive at the initial diagnosis of coeliac disease,1 7 most patients had negative coeliac disease-specific antibodies at the time of RCD diagnosis reflecting strict adherence to a GFD.13 15 17 Positive coeliac disease-specific serology can be present in 19–30% of patients with RCD despite good compliance to a GFD as assessed by dietitian interview; thus, positive coeliac disease-specific serology does not necessarily exclude the diagnosis of RCD.15 17 Although low-grade gluten contamination by a hidden source could be an issue,21 22 other non-dietary reasons for persistence of positive coeliac disease-specific serology in RCD could be: (1) coeliac disease-specific antibodies kinetics, (2) induction of tissue transglutaminase upregulation by severe inflammation or destructive lesions, and (3) coexistent autoimmune disorders associated with false-positive coeliac disease serology.15 23–25 A short period of close dietary surveillance is usually useful to clarify the origin of persistence of positive coeliac disease-specific serology but additional therapies or nutritional support should not be withheld if the clinical condition of the patient deteriorates.16 17
Endoscopic and imaging findings
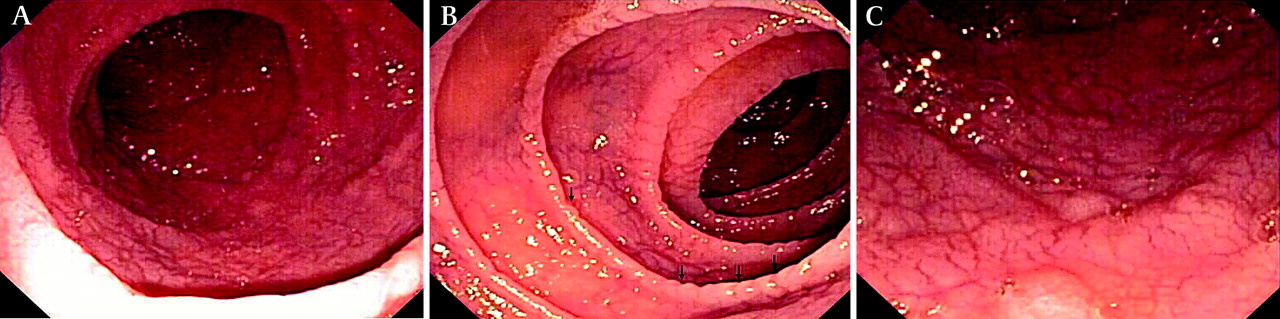
Standard upper gastrointestinal endoscopy and capsule endoscopy frequently show macroscopic features of villous atrophy or ulcerations 15 17 26 27 (figure 1).

Endoscopic abnormalities in refractory coeliac disease. Note the presence of classic endoscopic signs of villous atrophy such as loss of Kerckring's folds in the duodenum (A), scalloping of circular folds (arrows) (B), and fissuring with a mosaic pattern (C). These findings are not specific for refractory coeliac disease but excellent predictors of mucosal disease. (Courtesy of Dr Louis M. Wong Kee Song, Gastroenterology and Hepatology, Mayo Clinic, United States.)
However, erosions or ulcerations detected by capsule endoscopy in some patients with symptomatic, treated coeliac disease may not be always be related to RCD but to non-steroidal anti-inflammatory drug injury. Ulcerative jejunitis or large ulcerations (>1 cm) are common in patients with RCD type 2.15 17 Ulcerated nodular mucosa, occluding mass, or stricture suggest malignant complications.26–28 Double-ballon enteroscopy can efficiently detect or exclude ulcerative jejunitis or EATL in patients with RCD, especially when suggested by other imaging modalities such as abdominal CT scan.29
Non-specific intestinal abnormalities (eg, bowel-wall thickening or ‘malabsorption pattern’) or mesenteric lymphadenopathy are present in up to 50% of patients with RCD.17 Cavitating mesenteric lymph node syndrome characterised by cystic change in mesenteric lymph nodes with or without spleen atrophy is rare but characteristic of coeliac disease, often associated with RCD type 2.17 30 Small splenic volume (<122 cm3), intussusception, bowel wall thickening, and lymphadenopathy were more commonly detected by abdominal CT in patients with RCD type 2 or EATL as compared to uncomplicated coeliac disease or RCD type 1.31 18F-fluorodeoxyglucose positron emission tomography (PET) was more sensitive and specific than conventional CT for detection of EATL in patients with RCD, although the overall number of patients included in this prospective comparative study was small.32
Diagnosis
General diagnostic approach and differential diagnosis
The diagnosis of RCD requires a combination of clinical and pathological findings. Indeed, the diagnosis is made on the basis of strong evidence of coeliac disease, supplemented with systematic exclusion of both other causes of non-responsive coeliac disease or villous atrophy and malignancy. Although RCD is a diagnosis of exclusion, it is supported by objective findings in laboratory and histological studies. The availability of novel tests for detection of abnormal (clonal) intraepithelial lymphocytes in the intestine has facilitated the confirmation of RCD type 2 (figure 2).

Diagnostic approach in refractory coeliac disease.
The differential diagnosis of RCD includes other causes of villous atrophy associated with severe symptoms and/or refractory diarrhoea such as (not limited to) tropical sprue, collagenous sprue, adult-onset autoimmune enteropathy, hypogammaglobulinaemia and Crohn's disease.8 12 (table 1) Folic acid deficiency, a history of travel to endemic areas, and clinical response to a combination of folic acid and antibiotics supports tropical sprue as an alternative diagnosis.33 The presence of a subepithelial collagen deposition band together with clinical criteria of refractory diarrhoea is characteristic of collagenous sprue.34 Gut epithelial cell antibodies (either anti-enterocyte or anti-goblet cell) are important supportive evidence for the diagnosis of adult-onset autoimmune enteropathy.35
Major causes of villous atrophy in adults
Specific diagnostic approach
Confirming the diagnosis of coeliac disease
The first step in the evaluation of a potential case of RCD is to confirm whether the initial diagnosis of coeliac disease was correct.8 12 This requirement is easy to meet when patients had a combination of positive coeliac disease-specific serological tests, compatible histological findings in the intestinal biopsy, coeliac disease-permissive gene pairs encoding the human leucocyte antigens (HLA) DQ2 or DQ8, family history of coeliac disease, and medical history of unequivocal clinical or histological response to GFD.1 7 However, confirmation or exclusion of coeliac disease diagnosis can be challenging in some patients, especially those with a primary non-response to GFD.9 Practically all patients with coeliac disease carry DQA1*05.DQB1*02 encoding HLA-DQ2 or DQA1*03.DQB1*0302 encoding HLA-DQ8, thus the absence of either of these gene pairs has a very high negative predictive value for coeliac disease and should prompt consideration of other causes of refractory sprue.36–38 The presence of biopsy-proven dermatitis herpetiformis confirms the diagnosis of coeliac disease.39 Positive tissue transglutaminase (tTGA) or endomysial antibodies (EMA) at initial diagnosis of coeliac disease or at any time in the clinical course of the disease helps to confirm the diagnosis of coeliac disease because of their excellent specificities >99% when villous atrophy is present;40 however, most patients with developed RCD are likely to have negative tTGA and EMA.15 17Accordingly, negative coeliac disease-specific serology does not exclude the diagnosis of coeliac disease once the refractory state is fully developed. A family history of coeliac disease in first-degree relatives (especially siblings) further supports the diagnosis of coeliac disease in patients with compatible histology.37The diagnosis of coeliac disease relying only on histological findings or clinical improvement after GFD in the absence of other diagnostic criteria41 is not reliable because coeliac disease is just one of many causes of villous atrophy and either clinical response to GFD or exacerbation after gluten re-introduction may have an unacceptable low positive predictive value.34 42 43 Thus, a critical review of prior tests and especially histology slides is crucial to determine the accuracy of a prior diagnosis of coeliac disease. A history of travel to or residence in a location at risk for tropical sprue is also important in the identification of this readily treatable disorder.44A well-defined diagnosis of coeliac disease is clinically relevant to differentiate RCD from other more heterogeneous causes of non-coeliac disease associated refractory diarrhoea or enteropathy histologically indistinguishable from coeliac disease.12 35 The term ‘unclassified sprue’ has been used to designate individuals in whom the underlying malabsorptive disorder could not be adequately defined using all current diagnostic methods (box 1).9 12 28
The first step in the evaluation of a potential case of refractory coeliac disease (RCD) is to confirm whether the initial diagnosis of coeliac disease was correct
Negative coeliac disease-specific serology does not exclude the diagnosis of coeliac disease once the refractory state is fully developed
Human leucocyte antigen (HLA)-DQ status may be clinically relevant to exclude the diagnosis of coeliac disease
Critical review of prior laboratories studies including serology, pathology, and HLA-DQ status are crucial to confirm a prior diagnosis of coeliac disease
Aetiologies and diagnostic approach in ‘non-responsive’ coeliac disease
‘Non-responsive’ coeliac disease is a relatively common clinical scenario characterised by a lack of initial response to a GFD, or recurrence of symptoms or laboratory abnormalities typical of untreated coeliac disease while on a GFD in a patient who responded initially to the GFD.10 The aetiologies of non-responsive coeliac disease vary from centre to centre.10 11 45 Overt or inadvertent gluten contamination appears to be the most common cause of non-responsive coeliac disease seen in 36–51% of referral patients.10 11 Other aetiologies include microscopic colitis, small-bowel bacterial overgrowth, lactose intolerance and functional bowel disorders.8 10 11 45 (table 2) RCD is a diagnosis of exclusion that requires the elimination and/or treatment of other causes of non-responsive coeliac disease.
Aetiologies of non-responsive coeliac disease
While complete histological recovery after gluten exclusion may take several years in adults, clinical and serological responses are usually attainable after just a few weeks or months of gluten withdrawal, respectively.6 40 46 Thus, persistent or recurrent symptoms, positive coeliac disease-specific serology, and/or villous atrophy after 6–12 months on a GFD are atypical for uncomplicated coeliac disease and may require further evaluation. Persistently positive tTGA or EMA are suggestive of gluten contamination.11 Detailed evaluation of the strictness of the GFD by a skilled dietitian with special emphasis in revealing potential hidden sources of gluten is required.21 46 Other causes of symptoms on a GFD such as microscopic colitis, pancreatic insufficiency, small-intestine bacterial overgrowth, and irritable bowel syndrome require careful consideration prior to diagnosing RCD.8 Repeat intestinal biopsy may help to differentiate causes of non-responsive coeliac disease associated with ongoing villous atrophy (eg, gluten contamination, small-bowel bacterial overgrowth, RCD) from those associated with normal duodenal biopsy (eg, microscopic colitis, irritable bowel syndrome). Additionally, upper endoscopy permits the collection of duodenal fluid for culture of bacteria and extensive mucosal sampling to detect the presence of abnormal (clonal) T cells in the intestine. Persistent inflammation and villous atrophy after GFD without symptoms, while not traditionally considered as part of the RCD spectrum, may represent in a minority of patients a ‘latent’ form of RCD with a higher risk of development of symptomatic RCD or lymphoma over time.47 Finally, multiple conditions associated with persistent symptoms may occur together in the same patient.10 48 Thus, before a diagnosis of RCD could be taken as certain, a systematic investigation and exclusion of all other aetiologies of non-responsive coeliac disease is advisable.10 17 (box 2)
Overt or inadvertent gluten contamination appears to be the most common cause of non-responsive coeliac disease
Persistent or recurrent symptoms, positive coeliac disease-specific serology, and/or villous atrophy after 6–12 months on a gluten-free diet (GFD) are atypical and may require further evaluation
Refractory coeliac disease (RCD) is a diagnosis of exclusion that requires the elimination of other causes of non-responsive coeliac disease associated with villous atrophy
Exclusion of malignancy
The presence of fever, nocturnal diaphoresis, pruritus, significant unexplained weight loss, anorexia, overt or occult gastrointestinal bleeding, abdominal pain, and bowel obstruction do not occur in treated coeliac disease and suggest an underlying complication, especially ulcerative jejunitis or malignancies such as EATL and small-bowel adenocarcinoma.9 13 28 49 Subjects with EATL diagnosed before coeliac disease has been diagnosed should not be considered as affected by RCD because the outcome is determined by the neoplasm.49 This group has been arbitrarily classified as ‘primary’ EATL.13 Alternatively, ‘secondary’ EATL is used to describe the development of EATL after long-lasting well-controlled coeliac disease for several months or years before the diagnosis of RCD and subsequent EATL.13 15 17 50 Recently, two distinct groups of EATL were delineated morphologically and genetically; type 1 EATL, observed in 80–90% of cases, appears to be linked pathogenetically to coeliac disease and RCD. This subtype is characterised by non-monomorphic cytomorphology, CD56 negativity, and chromosomal gains of 1q and 5q.51 EATL is rare in the general population (incidence of 0.10 per 100 000 inhabitants per year in The Netherlands)52 but may occur in 60–80% of patients with RCD type 2 within 5 years and also has been occasionally described in patients with RCD type 1.13–15 17 The presence of intraepithelial γ/δ T lymphocytes is inversely correlated with lymphomagenesis in RCD.53 Extensive discussion of EATL treatment is beyond the scope of this review but regardless of EATL subtype, the prognosis is poor despite aggressive therapies, with a 5-year survival rate of 8–20%.13 49
Patients with coeliac disease have a higher risk of small bowel adenocarcinoma that may be a rare cause of deterioration of well-controlled treated coeliac disease.8 54 55 Small bowel adenocarcinoma in the context of coeliac disease is associated with better survival than the sporadic counterpart.56
Novel endoscopic or imaging techniques are available to visualise the entire small-bowel mucosa such as wireless capsule endoscopy, CT or MRI enterography, double or single balloon enteroscopy, and PET scan. These new techniques are of great utility to exclude malignancy in patients with possible RCD.1 8 13 15–17 29 42 55 However, the most cost-effective diagnostic approach is unclear because sensitivity and specificity of individual or combined findings with each diagnostic modality are unknown and comparative studies between methods are lacking.20 27 It is the authors' practice to use a combination of wireless capsule endoscopy that provides excellent luminal detail and CT–enterography to evaluate the wall of the intestine and extra-intestinal structures as initial screening tests to exclude malignancy in patients with RCD. Small-bowel enteroscopy is used to sample areas beyond the reach of standard endoscopy when the suspicion of malignancy is high. PET scan may be useful in patients with RCD type 2. Other diagnostic modalities are selected on a case-by-case basis.
Detection of abnormal intraepithelial lymphocyte phenotype
The detection of abnormal intraepithelial lymphocyte phenotype is the basis for subclassification of RCD and may also have prognostic significance.8 17 57 The current methods can be done in fixed (double CD3/CD8 immunohistochemistry, T cell receptor clonal rearrangement by PCR) or on fresh frozen intestinal tissue (flow cytometry).57–60 The abnormal phenotype is supported by (1) loss of normal surface markers CD3, CD4 and CD8 with preserved expression of intracytoplasmic CD3 (CD3ε) in >50% of intraepithelial lymphocytes as evaluated by immunohistochemistry or >20–25% as determined by flow cytometry (eg, CD103+, CD45+, CD7+, CD3−, CD8−); and (2) detection of T cell receptor chains (γ or δ) clonal rearrangement by PCR15 57–59 (figure 3). Defective synthesis or association of T cell receptor chains may explain the loss of surface TCR−CD3 expression in patients with RCD type 2 and EATL.61 While immunohistochemistry and PCR methods have been widely accepted, application of flow cytometry for detection of abnormal intraepithelial lymphocyte phenotype is relatively novel with promising results in a single centre59 (figure 4). Double CD3/CD8 immunohistochemistry have been proposed for initial evaluation of potential RCD because of the advantages of simplicity, accuracy in paraffin-embedded duodenal tissue, and low cost.58 T cell receptor clonal rearrangement by PCR is required to confirm the presence of clonal intraepithelial lymphocyte phenotype.15 17 57 58 62 It is the authors' practice to use both double CD3/CD8 immunohistochemistry and T cell receptor clonal rearrangement by PCR for the clinical evaluation of patients with clinical criteria of RCD because of the imperfect correlation between the two techniques.17 This approach is especially useful to detect RCD cases with a normal intraepithelial lymphocyte phenotype (CD3+CD8+) by immunohistochemistry but monoclonal T cell receptor rearrangement.63 Furthermore, the presence of concurrent persistent monoclonality and aberrant immunophenotype (especially if ≥80% CD3ε+CD8− intraepithelial lymphocytes) is a strong predictor of EATL development.64 Trisomy 1q22-44 has been observed in clonal intraepithelial lymphocytes characteristic of RCD type 2, thus, karyotypic studies may be helpful to categorise patients.15 65 Abnormal (clonal) intraepithelial lymphocytes may also be detected in the subepithelial layer, lamina propria, or even extra-intestinal locations such as skin, blood, bone marrow, liver, and bronchioloalveolar fluid in patients with RCD type 2, in the absence of overt EATL.15 66

Immunophenotype of intraepithelial lymphocytes in refractory coeliac disease (RCD) by CD3 and CD8 immunostaining. (A) Duodenal biopsy specimen from a patient with RCD type 1 (A1; H&E; magnification, ×20) showing partial villous atrophy and an increased number of intraepithelial lymphocytes with normal immunophenotype characterised by expression of CD3 (A2) and CD8 (A3). (B) Duodenal biopsy specimen from a patient with type 2 RCD (B1; H&E; magnification, ×20) showing villous atrophy and abnormal intraepithelial lymphocytes characterised by expression of (cytoplasmic) CD3 (B2) but mostly CD8− (B3). Brown colour denotes positive immunostaining (magnification, ×20). (Courtesy of Dr Tsung-Teh Wu, Anatomic Pathology, Mayo Clinic, United States.)

Example of flow cytometry analysis of intestinal lymphocytes isolated from duodenal biopsy specimens in a patient with refractory coeliac disease (RCD) type 2. Upper part: abnormal intraepithelial lymphocytes (92–93%). Lower part: abnormal lamina propria lymphocytes (57%). Left: lymphocyte selection gate based on CD45 positivity and low side scatter. Middle: the abnormal T-cell population by double staining of surface CD3- and CD7+ within the CD45+ cells. Right: the abnormal surface CD3−, cytoplasmic CD3+ cells by double staining shown within the CD7+ CD45+ cells. (Courtesy of Dr Wieke HM Verbeek, VU University Medical Center, Amsterdam, Netherlands.)
Clinical classification and prognosis
The clinical classification of RCD is based on the immunophenotype of intraepithelial lymphocytes and supported by clinical evolution (table 3). Abnormal (clonal) intraepithelial lymphocytes are the hallmark of RCD type 2. RCD type 2 is associated with poor prognosis despite conventional therapeutic intervention with 5-year survival rates of 40–58%.13 15–17 Poor prognosis is largely explained by the much more frequent progression to overt EATL in patients with RCD type 2 (figure 5).13 15 17 50 57 67 Recent evidence suggests that continual monitoring of both immunophenotype and clonality of intraepithelial lymphocytes may be more accurate than snapshot analysis for both correct ascertainment of RCD subtype and prediction of risk of lymphomagenesis.64 The prognosis of RCD type 1 is much better as compared to RCD type 2 but the rates of complications and mortality appear to be much higher than those observed in non-complicated coeliac disease.14 16 17 Besides RCD subtype and lymphoma development, other prognostic factors appear to be important such as older age at RCD diagnosis (eg, age ≥65 years), albumin ≤3.2 g/dl, haemoglobin ≤11 g/dl, and total villous atrophy at RCD diagnosis.17 A new clinical staging model for RCD based on the cumulative effect of clinical prognostic factors has been proposed using single centre data but requires further validation before being ready for clinical use.17
Differential diagnosis of refractory coeliac disease type 1 and refractory coeliac disease type 2

Histological features of enteropathy-type T cell lymphoma. (A) Low-power magnification of surgical specimen from the jejunum showing subtotal villous atrophy and diffuse infiltration by enteropathy-type T cell lymphoma in a patient with long-lasting refractory coeliac disease (RCD) type 2 (H&E; magnification, ×4). (B) Detail of a monotonous tumour cell population with rounded vesicular nuclei, single nucleolus, and abundant cytoplasm (H&E; magnification, ×40). (Courtesy of Dr Tsung-Teh Wu, Anatomic Pathology, Mayo Clinic, United States.)
Treatment
Evidence for treatment of RCD is based on case reports, open-label observational or prospective experiences, and expert opinion.8 20 68 There are no randomised clinical trials. The lack of data is partly explained by the rarity of RCD. The paucity of data should be recognised when making therapeutic decisions in patients with RCD.17 Furthermore, as the diagnostic criteria for RCD have changed over time, historical reports on treatment need to be interpreted cautiously because the absence of a clear distinction between RCD and other causes of refractory sprue as well separating RCD type 1 and type 2.
Dietary and nutritional assessment
Hospitalisation is sometimes necessary to monitor adherence to the GFD and for treatment of severe nutritional complications or dehydration. Total parenteral nutrition was necessary in 28–60% of patients with RCD because of severe weight loss, malnutrition, multiple nutritional deficiencies, and severe hypoproteinaemia and/or steatorrhoea.15–17 Nutritional treatment should also include correction of trace element deficiencies, including zinc and copper, and addressing metabolic bone disease.46 55 The GFD reduces overall morbidity and mortality in coeliac disease;69 thus, although the benefit of a GFD in RCD is unknown, strict adherence to the GFD has been widely recommended.13 15–17 A GFD alone may be an effective maintenance therapy in exceptional cases. An elemental diet showed promising results in a small heterogeneous group of patients with refractory enteropathy without clonal intraepithelial lymphocyte phenotype.70
Alternative therapies
Prednisone (0.5–1 mg/kg/day), budesonide (9 mg/day), or a combination of prednisone and azathioprine (2 mg/kg/day) are clinically effective for inducing clinical remission and mucosal recovery in most patients with RCD type 1.13 15–18 57 68 71 72 Clinical response to steroids is observed in the majority (∼75%) of patients with RCD type 2; however, mucosal recovery is infrequent and progression to EATL is not prevented.13 15 17 Steroid dependence is observed in most patients with RCD type 1 or type 2.15 17 Clinical response to budesonide in RCD is clinically attractive because of its topical effect, extensive first-pass metabolism, good tolerability, and low frequency of serious adverse events after short-term use.17 18 73 Data on tolerability and safety after long-term use of budesonide in RCD are lacking but budesonide was well tolerated after 6 months of use for the maintenance of clinical remission in collagenous colitis.74 Clinical and histological improvement was observed in up to 61% of patients with RCD after open-label treatment with oral cyclosporine (5 mg/kg/day).75 Other immunosuppressive drugs or biological modifiers have been used with some clinical benefit in steroid-dependent or steroid-refractory patients including azathioprine, ciclosporin, infliximab (5 mg/kg/day), and alemtuzumab (30 mg twice a week per 12 weeks), though publication of successfully treated cases rather than failures must be considered (publication bias).76–80 In a small open-label pilot study, recombinant human interleukin 10 administrated subcutaneously (8 μg/kg three times a week per 3 months) was not effective to restore villi in the majority of patients with RCD.81 Although valuable for treatment of RCD type 1, azathioprine should be used with caution because of potential severe side effects, especially myelosuppression, infections and lymphomagenesis.82 83 Lymphomagenesis is of special concern in patients with RCD type 2 because of the higher risk of EATL development in this subgroup and the fact that monoclonality persists after treatment with azathioprine.68 71 76 Intravenous cladribine (0.1 mg/kg/day for 5 days) was well tolerated in an open-label study in patients with RCD type 2 previously treated with prednisone and/or azathioprine and can induce clinical improvement (36%), histological improvement (59%), and significant decrease in the number of clonal intraepithelial lymphocytes (35%).84 However, up to 41% developed EATL and died despite cladribine therapy.84 Case reports showed that alemtuzumab (anti-CD52 monoclonal antibody) or a combination of pentostatin (4 mg/m2 every 2 weeks per 24 weeks) and budesonide could similarly induce clinical and histological response as well as a decrease but not disappearance of clonal intraepithelial lymphocytes.79 80 85 The effect of drug-induced T cell clone suppression on lymphomagenesis is not completely understood but EATL may still occur over time in patients with RCD type 2 even after the T cell clone was not further detected in the intestinal tissue by conventional methods.17 Accelerated lymphomagenesis is a concern with cladribine and alemtuzumab, although the number of cases with EATL progression was small.15 84 Finally, high-dose chemotherapy followed by autologous haematopoietic stem cell transplantation (ASCT) has been explored for RCD type 2 in a pilot study from a single centre.86 The outcome of ASCT is disappointing for patients with overt EATL.17 87 For future studies, interleukin 15 blockade may represent one promising option for RCD type 2 because of the key role played by this cytokine in the disruption of intraepithelial lymphocyte homeostasis and lymphomagenesis.68 88
Surgery
The role of surgery in RCD is limited to the management of complications such as perforation, massive haemorrhage, high-grade obstruction, and cancer.13 15 Long-term clinical remission on a GFD alone after complete resection of ulcerative jejunitis has been reported, particularly if ulceration is localised to one part of the intestine16 28 (figure 6). The role of diagnostic laparotomy in RCD is now limited by the introduction of novel endoscopic modalities that permit visual evaluation and biopsy specimen sampling of the whole intestine such as single or double-balloon enteroscopy.29

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Histological findings in ulcerative jejunitis. Extensive ulceration (left) and villous atrophy (upper right) in the jejunum of a patient with refractory coeliac disease compatible with ulcerative jejunitis (A) (H&E; magnification, ×10). Most intraepithelial lymphocytes in the atrophic mucosa adjacent to ulceration express (cytoplasmic) CD3 (B) but not CD8 (C). Brown colour denotes positive immunostaining (magnification, ×20). (Courtesy of Dr Tsung-Teh Wu, Anatomic Pathology, Mayo Clinic, United States.)
Summary
Refractory coeliac disease (RCD) is defined by persistent or recurrent malabsorptive symptoms and villous atrophy despite strict adherence to a gluten-free diet (GFD) for at least 6–12 months in the absence of other causes of non-responsive treated coeliac disease and overt malignancy
The real prevalence of RCD is unknown but is probably rare
RCD may be classified as type 1 (normal intraepithelial lymphocyte phenotype), or type 2 (defined by the presence of abnormal (clonal) intraepithelial lymphocyte phenotype)
Several alternative therapies are beneficial in RCD type 1 especially prednisone, budesonide or a combination of prednisone and azathioprine but there are no established treatments for RCD type 2
RCD is associated with a higher risk of complications and mortality, especially RCD type 2 with a 5-year survival rate of 40–58%
Chemotherapeutic drugs alone or high-dose chemotherapy followed by autologous haematopoietic stem cell transplantation (ASCT) have been systematically explored for selected patients with RCD type 2 in a single centre. Further studies in other centres or probably as a multicentre effort are needed
Advances in basic science may be translated into future novel therapies, such as interleukin 15 blockade either as a single agent or combination therapy
Acknowledgments
The authors thank Deborah I. Frank for her assistance in the preparation of this manuscript.
References
Footnotes
Linked articles 186007
Funding This article was supported by the National Institutes of Health (NIH) under Ruth L. Kirschstein National Research Service Award/Training Grant in Gastrointestinal Allergy and Immunology (T32 AI-07047) (to AR-T) and the NIH grant DK-57892 (to JAM).
Competing interests Dr Murray is a consultant for ActogeniX Inc., Flamentera Inc., Ironwood Pharmaceuticals, and Alvine Inc. He is also an investigator for ALBA Therapeutics. Dr Rubio-Tapia declares no competing interest.
Provenance and peer review Not commissioned; externally peer reviewed