Article Text

Abstract
Background and aims: Treatment with infliximab induces remission in about 70% of patients with steroid refractory Crohn's disease. Because Crohn's disease is considered to be mediated by uncontrolled activation of mucosal T lymphocytes, we hypothesised that infliximab could induce apoptosis of T lymphocytes.
Methods: Induction of apoptosis in vivo was studied in 10 patients with therapy refractory Crohn's disease. In vitro, resting or stimulated Jurkat T cells were incubated with infliximab.
Results: Infusion of infliximab (5 mg/kg) in steroid refractory patients with Crohn's disease induced a clinical response in 9/10 patients but did not influence expression of activation markers, homing receptors, memory cells, Fas expression, or Bax/Bcl-2 expression on peripheral blood T lymphocytes. In contrast, a significant increase in CD3 and TUNEL positive cells within colonic biopsies was detected 24 hours after infusion of infliximab, suggesting that infliximab stimulates apoptosis of activated T lymphocytes but not of resting T cells. To test this hypothesis, the effects of infliximab on Jurkat T cells were investigated. We observed that infliximab induced apoptosis and an increase in the Bax/Bcl-2 ratio of CD3/CD28 stimulated Jurkat T cells but not of unstimulated Jurkat cells.
Conclusions: Our data indicate that infliximab treatment causes a rapid and specific increase in apoptosis of T lymphocytes in the gut mucosa. These findings may explain the rapid and sustained therapeutic effects of infliximab in Crohn's disease.
- apoptosis
- T lymphocytes
- Crohn's disease
- infliximab
- tumour necrosis factor
- TNF-α, tumour necrosis factor α
- CRP, C reactive protein
- CDAI, Crohn's disease activity index
- MFI, mean fluorescence intensity
- IFN-γ, interferon γ
Statistics from Altmetric.com
- TNF-α, tumour necrosis factor α
- CRP, C reactive protein
- CDAI, Crohn's disease activity index
- MFI, mean fluorescence intensity
- IFN-γ, interferon γ
Although the cause of Crohn's disease is unknown, substantial experimental and clinical evidence suggests that uncontrolled T lymphocyte activation is a central pathogenic mechanism.1,2 In active Crohn's disease, many proinflammatory cytokines are released within the mucosal compartment, of which tumour necrosis factor α (TNF-α) seems to have a particularly important role.3,4 The number of TNF-α secreting mucosal cells as well as stool TNF-α protein concentration are increased in Crohn's disease.5,6 A significant minority of Crohn's disease patients are refractory to standard medical treatment, consisting of corticosteroids, azathioprine, or methotrexate. Treatment of therapy refractory Crohn's disease patients with the human-mouse chimeric TNF-α neutralising antibody, infliximab, resulted in a therapeutic response in 65% of patients, and complete remission was induced in 33% compared with 17% and 4%, respectively, in placebo treated patients.7 In patients with enterocutaneous fistulas, infliximab treatment induced rapid closure in 55%, which was significantly better than the 13% closure in Crohn's disease patients on standard medical treatment.8 Several features distinguish the therapeutic effects of infliximab treatment from those of standard current medical therapy. The therapeutic effect of infliximab treatment can be observed almost immediately after infusion, with a reduction in C reactive protein (CRP) concentrations and normalisation of intravascular thrombin formation within a few days, and the effects of a single infusion are sustained for 8–12 weeks.9,10 Infliximab causes rapid suppression of mucosal inflammation, as indicated by a reduction in the number of lamina propria cells producing TNF-α and the chemokines RANTES and MIP-1α.11,12 Moreover, in contrast with current medical therapy, infliximab treatment has been demonstrated to consistently heal the primary lesion of Crohn's disease, the mucosal ulcer.13
These observations suggest that the effects of infliximab treatment are mediated by mechanisms that differ from current medical therapy. Unravelling of these mechanisms may shed light on the pivotal pathogenic mechanisms in Crohn's disease and direct novel intervention strategies.
Because the intestinal mucosa is exposed to a very high antigenic pressure, downregulation of T lymphocyte activation is an important regulatory mechanism within the mucosal compartment. Three mechanisms of peripheral T lymphocyte tolerance have been characterised: anergy, cytokine (interleukin 10 and transforming growth factor β) dependent downregulation, and activation induced programmed cell death (apoptosis).14 T lymphocyte apoptosis is importantly dependent on the intracellular concentrations of two proteins that control the mitochondrial ANT pore, Bax (pro-apoptotic)15 and Bcl-2 (anti-apoptotic).16,17 Hence, the Bax/Bcl-2 ratio is a proximal control switch that determines whether a T lymphocyte will respond to apoptotic signals.18,19 Indeed, resistance of lamina propria T lymphocytes to Fas mediated induction of apoptosis in Crohn's disease has recently been demonstrated to correlate with a markedly reduced intracellular Bax/Bcl2 ratio.20,21
Because we considered it unlikely that the rapid and sustained effects of infliximab treatment are solely explained by neutralisation of soluble TNF-α, we hypothesised that in addition to neutralising soluble TNF-α, the antibody would also affect activation of mucosal T lymphocytes, in particular their susceptibility to apoptosis. In the present study we have investigated induction of apoptosis of mucosal T lymphocytes in patients treated with infliximab, and sought a possible mechanism of action by stimulating Jurkat T lymphocytes in the presence of infliximab ex vivo.
MATERIALS AND METHODS
Patients
Ten consecutive patients with active Crohn's disease were included (mean age 28.6 years (range 21–38)). Mean disease duration was 7.5 (2.7) years (range 4–12) and mean Crohn's disease activity index (CDAI) was 404 (range 182–808). Localisation of disease activity was assessed by endoscopy (sigmoid and rectum (n=5), left colon (n=2), neoterminal ileum (n=3)). Treatment with corticosteroids for at least four weeks (10 mg/day, orally) had failed in all patients. Concomitant treatment with other immunosupressive drugs was allowed but the dosage had to be stable for at least eight weeks before enrolment. Other exclusion criteria were a positive human immunodeficiency virus test result or treatment with a drug that could potentially modulate TNF synthesis (thalidomide, pentaxivaline, and embrell). The local ethics committee granted approval for the study and all patients gave informed consent.
Treatment and assessments
A single intravenous dose (5 mg/kg) of the anti-TNF chimeric antibody (infliximab; Centocor, Malvern, Pennsylvania, USA) was administered as a two hour infusion. Patients were admitted to hospital for 24 hours and follow up was one week and six weeks after the initial infusion. Blood pressure, temperature, and pulse were measured before infusion. Thirty minute serial determinations were made after starting the infusion. Routine haematology, blood chemistry, and FACScan analysis were performed at 0, 2, 4, 8, 12, and 24 hours and at week 1 and week 6. Endoscopy was performed immediately before infusion and 24 hours later, and biopsies were taken from involved mucosa and not from ulcers. Location was verified by videotaping the entire procedure so that biopsies obtained 24 hours after the start of the treatment were taken at the same location as immediately before treatment. Patients used a daily diary system before and during the six week study period for assessment of CDAI. CDAI scores were determined within one week before infusion and after infusion at weeks 1 and 6. Clinical response was defined as a decrease in CDAI by 70 points or more, or induction of complete remission defined as a CDAI <150 points.
Cell culture and stimulation
The human Jurkat T cell line was cultured in RPMI 1640 (Bio Whittaker, Belgium) containing 10% fetal calf serum and the antibiotic ciprofloxacin (10 μg/ml), and subcultured twice a week. Cells were stimulated (concentration 1×106 cells/ml) in RPMI 1640 supplemented with 5% pooled human serum (Bio Whittaker). T cells were unstimulated or stimulated with soluble anti-CD3 and anti-CD28 antibodies (2 ng/ml; CLB, Amsterdam, the Netherlands) and incubated with anti-TNF antibodies (10 μg/ml, infliximab; Centocor, Malvern, USA) or its isotype matched control. In parallel experiments, cells were incubated with mouse antihuman anti-CD95 antibody (IgG 1 μg/ml; CLB).
TUNEL assay
The cell pellets of Jurkat cells were washed and cytocentrifuged on superfrost slides in phosphate buffered saline (pH 7.4) supplemented with 1% bovine serum albumin (Sigma Chemical Co, St Louis, Missouri, USA). Slides were air dried and stored at −20°C until use. Human intestinal mucosal biopsies were embedded in Tissue-tek (Sakure Finetech Europe BV, Zoeterwoude, the Netherlands), subsequently snap frozen in liquid nitrogen, and kept at −70°C until use. Frozen sections (6 μm) were cut on a cryostat and kept at −20°C until use. TUNEL assay was performed according to the manufacture's instructions (Boehringer Mannheim, Germany). In short, terminal deoxynucleotidyl transferase was used to label the DNA strand breaks with modified fluorescein labelled nucleotides. For light microscopy detection, the incorporated fluorescein was detected by antifluorescein antibody conjugated with AP. After substrate was added, slides were analysed by microscopy and image analysis to quantitate the staining.
Immunohistochemistry
Slides with 6 μm thick human intestinal biopsy sections were fixed with 1% paraformaldehyde, and mouse-antihuman CD3 (CLB) was used. Goat-antimouse-AP labelled antibody was used as a secondary antibody. Detection was done using Fast Red (Dako, Glostrup, Denmark).
Flow cytometry
Jurkat cells were washed twice after stimulation. Cells were permeabilised using lysing and permeabilising solution (Becton Dickinson, Rutherford, New Jersey, USA). Antibodies used were antihuman-Bax (Immunotech, Marseille, France) and antihuman-Bcl-2 (Dako). After addition of the primary antibody, cells were incubated for 30 minutes on ice and washed twice in cold FACS buffer (phosphate buffered saline containing 1% bovine serum albumin, 0.3 mmol/l EDTA, 0.01% sodium azide). Subsequently, cells were incubated with a goat-antimouse-FITC labelled antibody for another 30 minutes on ice. After two washes, cells were analysed by FACScan (Becton Dickinson) and 10 000 cells were counted. After correcting for control IgG fluorescence, specific antibody binding was expressed as mean fluorescence intensity (MFI). Bax/Bcl2 ratios are expressed as MFI. During the six week period, blood samples of patients and six healthy volunteers (mean age was 31 years (range 26–43), four females, two males) were analysed for several markers.
Erythrocytes were lysed with ice cold isotonic NH4Cl solution (155 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA, pH 7.4) for 10 minutes. Cells were centrifuged at 600 g for five minutes at 4°C. The remaining cells were washed twice with cold FACS buffer. For staining, 1×106 cells/well (96 well microplate; Greiner BV Labor Techniek, Alphen aan de Rijn, the Netherlands) were incubated with the following fluorescent labelled mouse monoclonal antibodies against humans: CD4, CD3, CD95, CD134, CD154, Bax, CD45RO, CD45RA (Immunotech), CD25, CD8, (CLB), Bcl2 (Dako), A4B7, CD120a, and CD120b (Sanbio). After addition of the primary monoclonal antibody to the cell suspension, the cells were incubated for 30 minutes at 4°C and washed twice in FACS buffer. TNF binding potential was measured using a FITC labelled human TNF-α. The appropriate isotype controls were included in all experiments. Lymphocytes were gated by forward and side scatter using a FACScan flow cytometer in conjunction with the FACScan software (Becton Dickinson, Mountain View, USA). A total of 5000 cells were counted. Results are expressed as the percentage of gated cells positive for the monoclonal antibodies used or as MFI after subtraction of control IgG fluorescence.
Interferon γ production by activated Jurkat cells
Interferon γ (IFN-γ) levels in Jurkat supernatants were measured by ELISA according to the manufacturer's instructions (CLB).
Detection of anti-DNA antibodies
Sera were tested for DNA binding using an immunofluorescence technique on Crithidia luciliae.22,23 As FITC conjugates, antibodies to human Ig (SH17-01-F5; diluted 1/100), to human IgG (KH16-107-F3; 1/100), and to human IgM (KH15-F1; 1/50), all from CLB, were used.
Statistics
For the in vitro Jurkat experiments, statistical analysis was performed using the Wilcoxon test in SPSS for Windows, version 7.5. For other experiments and the clinical study results, ANOVA and the Student's t test were performed where appropriate.
RESULTS
Clinical outcome of infliximab treatment of Crohn's disease patients
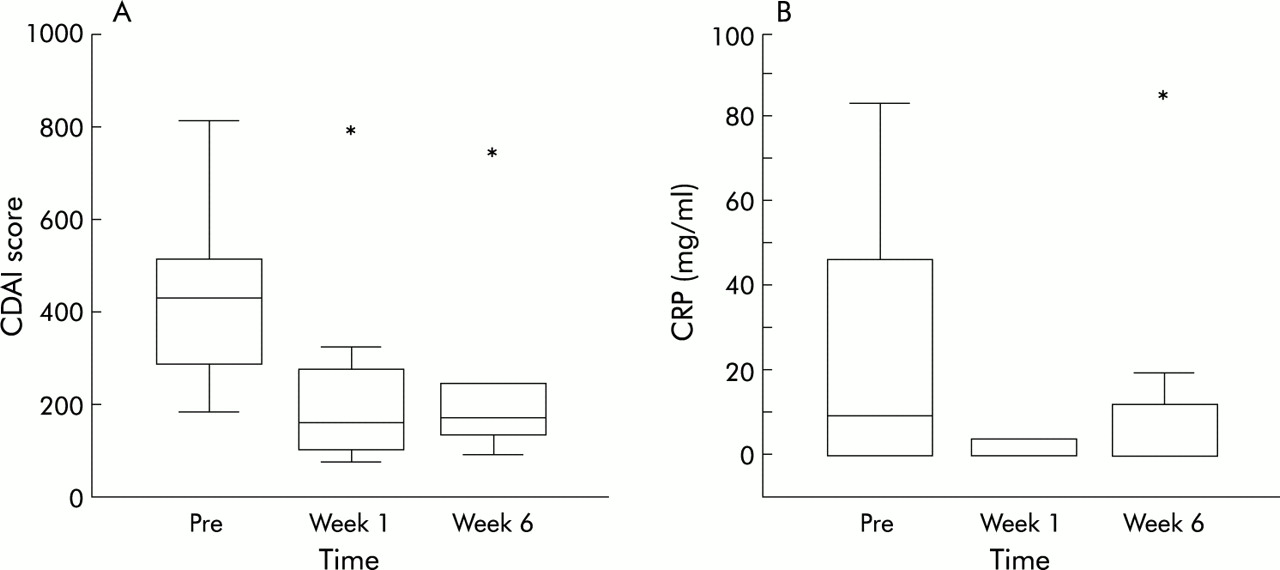
Ten patients with severe steroid refractory Crohn's disease were treated with a single intravenous dose (5 mg/kg) of infliximab. Infusion of infliximab was well tolerated and no febrile or allergic reactions were observed. Nine patients had a clinical response, as defined as a reduction in CDAI of 70 points or more.7 The one non-responding patient had an ileal-rectal anastomosis and therefore a short bowel high throughput syndrome. For this reason the number of stools, the main indicator in CDAI, remained equally high after treatment with infliximab. However, this patient did respond in terms of induction of apoptosis (see below). The nine responding patients reported improvement in subjective symptoms within one week after the start of treatment. The mean CDAI was 404 before infusion and decreased to 240 after one week and to 262 after six weeks (fig 1A). One patient relapsed five weeks after infusion but in the eight remaining patients responses were sustained during the six weeks of follow up. CRP concentrations normalised within one week in all patients who had increased levels before infusion and remained low throughout the study period (fig 1B). In the single relapsing patient, CRP levels rose towards preinfusion levels six weeks after infusion. Two hours after the start of infusion, peripheral blood lymphocyte counts slightly decreased in 7/10 patients (fig 2) and a transient relative lymphocytosis was observed in all patients at 24 hours (p<0.05). Lymphocyte counts returned to baseline in all patients at six weeks.
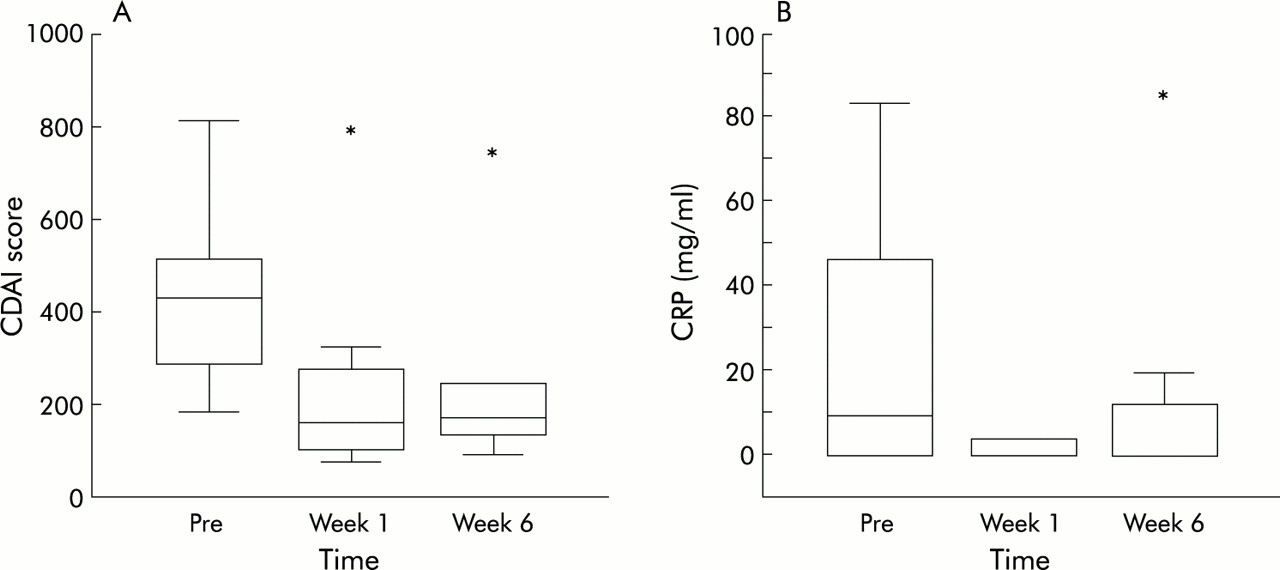
Clinical responses. (A) Boxplot of the Crohn's disease activity index (CDAI) score of the 10 treated patients before (Pre) and one and six weeks after intravenous infusion of infliximab. *Non-responding patient. (B) Boxplot of C reactive protein (CRP) concentrations measured during the study period. *Relapsing patient.

Total number of lymphocytes throughout the study period. Values are mean (SEM).
Infliximab treatment induces anti-dsDNA antibodies
Sera of all patients were tested for DNA binding using an immunofluorescence technique on Crithidia luciliae.22,23 At study entry, none of the patients showed anti-DNA antibodies. Six weeks after infusion of infliximab, four of 10 patients became positive for IgM anti-DNA antibodies.
No change in peripheral T lymphocyte markers after infliximab treatment
Peripheral T lymphocytes were frequently analysed throughout the study period for changes in expression of various membrane activation markers and intracellular Bax/Bcl-2 levels. No significant changes over time were noted for expression of activation markers (CD25, CD134, CD154), homing receptor (α4β7), memory cells (CD45RO, CD45RA), Fas expression (CD95), or Bax/Bcl-2 ratio. Expression of most of these markers was not different from normal healthy controls. However, compared with controls, baseline percentage of CD134 (patients 37.4%, controls 12.6%; p<0.05) and percentage of CD95 (patients 69.2%, controls 40.1 %; p<0.05) positive T lymphocytes was increased in patients with Crohn's disease. Infliximab treatment did not alter expression of these markers (table 1). Expression of both TNF receptors (p55 and p75) was similar in patients and controls but the TNF binding capacity of lymphocytes and granulocytes, as measured by FACScan analysis, was significantly decreased in patients with active Crohn's disease (table 1). Infliximab treatment caused no significant change in the TNF binding capacity of lymphocytes or granulocytes during the six week study period. Hence the beneficial effects of infliximab treatment of active Crohn's disease were not reflected in major changes in properties or apoptosis of circulating T lymphocytes.
Differences in CD95 and CD134 expression (%), and tumour necrosis factor (TNF) binding potential
Infliximab induces apoptosis of CD3 positive lamina propria T lymphocytes
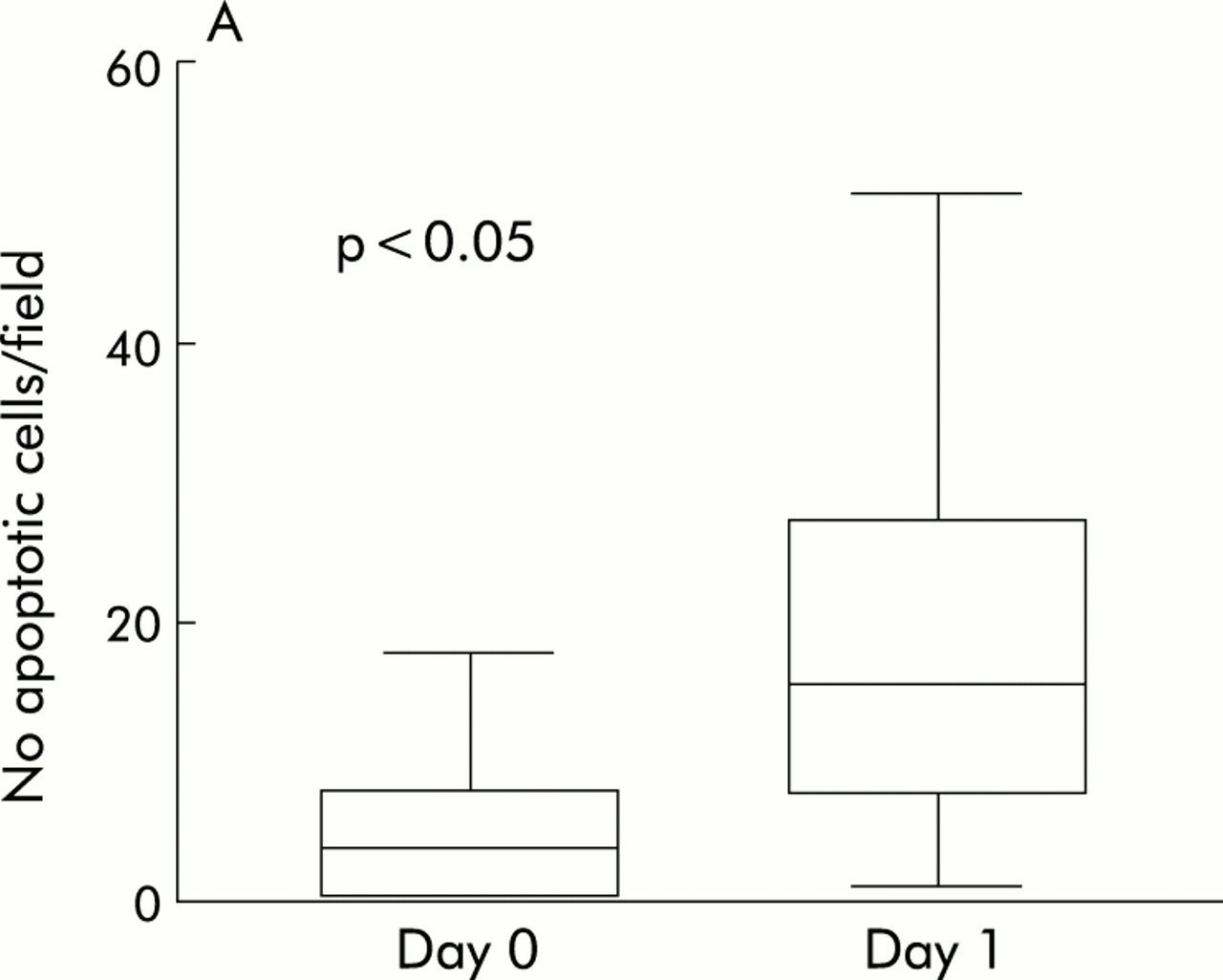
Subsequently, we investigated whether cells present at the site of active disease were a target of infliximab induced apoptosis. To this end, the intestinal biopsies taken during endoscopies performed immediately before and 24 hours after infliximab infusion were analysed for apoptotic cells using the TUNEL assay. A quantitative image analysis method was used to express the number of apoptotic cells as a fraction of the total number of lamina propria cells. The total number of apoptotic cells in the biopsies of 10 consecutive patients was increased 24 hours after the start of treatment. Quantitative analysis indicated that the number of apoptotic cells increased from 5.4 cells/field to 22.9 cells/field (fig 3A). To investigate which cells underwent apoptosis, serial tissue sections of two patients were stained for TUNEL positive and CD3 positive cells. Analysis of these serial sections indicated that the majority of apoptotic cells were lymphocytes (fig 3B). We concluded that although infliximab has little influence on resting circulating T cells, it is an active promoter of CD3 positive T lymphocyte apoptosis in the lamia propria.


Infliximab induces apoptosis of CD3 positive T lymphocytes in the lamina propria of treated patients. (A) Boxplot of total number of apoptotic cells measured in biopsies taken before and 24 hours after treatment. (B) Immunohistochemical staining of two consecutive slides of a biopsy taken one day after treatment of one patient. The TUNEL positive cells (left) are located on the same site as the CD3 positive cells (right).
Infliximab induces apoptosis of CD3/CD28 double stimulated Jurkat cells
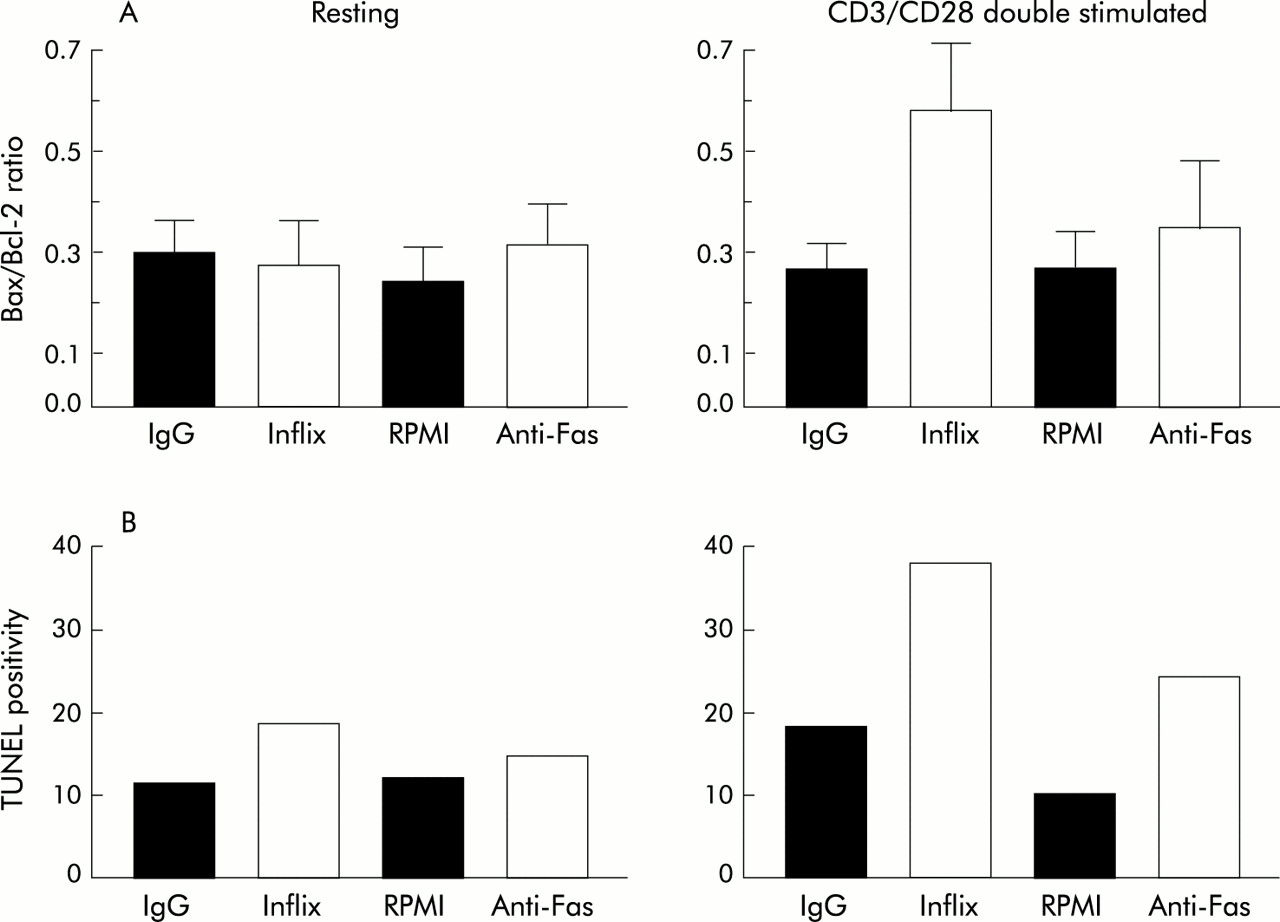
To test the hypothesis that infliximab is a specific inducer of programmed cell death of activated but not resting T lymphocytes, we determined the ability of infliximab to induce apoptosis of CD3/CD28 stimulated or resting Jurkat T cells. In resting cells, infliximab did not change the Bax/Bcl-2 ratio, but at four and 24 hours after CD3/CD28 stimulation, a significant increase in the Bax/Bcl-2 ratio was found compared with the isotype control (respectively from 0.27 to 0.43 (p<0.05) and from 0.26 to 0.59 (p<0.05)) (fig 4).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effects of infliximab on Jurkat T cells. (A) Bax/Bcl2 ratio in Jurkat cells determined by FACScan analysis. Data are expressed as mean (SEM) of six independent experiments. Jurkat cells were incubated for 24 hours with IgG isotype control, infliximab (Inflix), RPMI, and anti-Fas antibody. Cells were either not stimulated (left) or stimulated with anti-CD3/anti-CD28 antibodies (right). (B) Quantitative results of a TUNEL assay on cytospins of Jurkat cells. TUNEL positive cells were counted in non-stimulated (left) and stimulated (right) cells.
Both Fas antibody and infliximab increased the number of apoptotic cells, as assessed by TUNEL assay (fig 4), in an activation dependent manner. Hence although both infliximab and Fas antibody induced apoptosis of activated Jurkat T lymphocytes, only after infliximab exposure did this seem to be mediated by an increase in the Bax/Bcl-2 ratio.
IFN-γ levels were measured in the supernatant of Jurkat cells 48 hours after stimulation with CD3/CD28. After incubation with infliximab, production of IFN-γ significantly decreased. In stimulated cells, mean IFN-γ concentration was 116.0 (37.3) pg/ml compared with a mean of 28.9 (6.5) pg/ml (p<0.05) in CD3/CD28 stimulated cells that were incubated with infliximab. When T Jurkat cells were incubated with another known inducer of apoptosis, a similar reduction in IFN-γ production was found (mean 18.4 (4.9) pg/ml).
Together, these experiments confirm that infliximab is a direct inducer of apoptosis in activated but not resting T lymphocytes. We propose that such specific apoptosis of activated T lymphocytes mediates the long term effects of infliximab in Crohn's disease.
DISCUSSION
In the present study, a single infusion of infliximab resulted in a rapid clinical response in 9/10 patients with severely active Crohn's disease. Activation markers and intracellular Bax/Bcl-2 concentrations were frequently assessed during the six week study period using FACS analysis of peripheral blood mononuclear cells but no changes were observed. In contrast, in intestinal biopsies a significant increase in the number of apoptotic cells was found 24 hours after treatment, and by analysis of serial sections the vast majority of TUNEL positive cells were found to be CD3 positive. Furthermore, the in vitro experiments with Jurkat cells suggest that this effect is mediated by a change in the Bax/Bcl-2 ratio whereas the functional consequences of infliximab treatment were reflected in a significant reduction in IFN-γ production by stimulated Jurkat cells. Hence our data indicate that infliximab treatment can induce apoptosis of activated but not resting Jurkat T lymphocytes, and that infliximab treatment of Crohn's patients causes a rapid and specific increase in the number of apoptotic T lymphocytes in the gut mucosal compartment, but not of peripheral blood mononuclear cells.
It has been reported previously that infliximab can cause complement dependent lysis of mononuclear cells24 but it is unlikely that this mechanism occurs in vivo. Firstly, infusion of infliximab is well tolerated and patients do not develop symptoms of a cell lysis syndrome. Secondly, in contrast with the effects of CD4 depleting antibodies, no significant decrease in peripheral blood mononuclear counts occurred after infliximab treatment.25 Indeed, as has been reported in rheumatoid arthritis patients, infliximab caused a transient lymphocytosis, which may be a result of acute inhibition of T lymphocyte trafficking to areas of inflammation.26 We interpret our results to indicate that the effects of infliximab in patients with Crohn's disease are secondary to specific induction of apoptosis of lamina propria T lymphocytes. Apoptosis resistance of T lymphocytes has been implicated as a pathogenic mechanism in several immune mediated diseases, and it has been speculated that such a mechanism may constitute a genetic basis for multiple sclerosis.27 In Crohn's disease, mucosal T lymphocytes are less susceptible to Fas mediated apoptosis, which coincides with an increased Bcl-2 concentration.20,21 It is uncertain whether Crohn's disease patients have a genetic defect in activation induced T lymphocyte apoptosis, or whether the increased Bcl-2 concentration within mucosal T lymphocytes is a result of mucosal inflammation. Indeed, cytokines that activate the interleukin 2 common γ chain (interleukin 2, interleukin 7) are known to increase Bcl-2 concentrations within T lymphocytes.28,29
In the search for a mechanism of action, we have extended our clinical study with several ex vivo experiments. We found that incubation of CD3/CD28 stimulated Jurkat T lymphocytes incubated with infliximab caused a significant reduction in IFN-γ production. Because infliximab does not recognise IFN-γ, an effect on T lymphocyte survival was suspected. Indeed, very similar effects on CD3/CD28 stimulated IFN-γ production were observed when methotrexate, which is known to induce T lymphocyte apoptosis, was added. Indeed, using TUNEL assays, it became clear that infliximab caused significant induction of activation induced apoptosis, which was associated with an increase in the Bax/Bcl2 ratio.
Although we have found that binding of infliximab to activated Jurkat cells results in a change in the Bax/Bcl-2 ratio, the precise mechanism of induction of apoptosis remains unclear, and it may seem paradoxical that neutralisation of TNF-α, a cytokine that is involved in apoptosis induction in certain types of cells, leads to apoptosis of T lymphocytes. On the other hand, TNF-α also induces the transcription factor nuclear factor κB that has strong antiapoptotic effects in T lymphocytes.30 Hence it is conceivable that neutralisation of soluble TNF-α would reduce nuclear factor κB activation which in activated T lymphocytes is known to protect against apoptosis induction, leaving the cells exposed to induction of apoptosis through Fas/Fas ligand interactions. Infliximab binds to membrane expressed TNF-α that has an unusually long intracellular tail, but is not thought to contain known signal transducing domains. None the less, it is possible that engagement of transmembrane TNF-α may cause “reverse signal transduction” through outside-in signal transduction. It is conceivable that such a mechanism results in a change in the Bax/Bcl-2 ratio, either directly by interfering with upstream elements of the apoptosis pathways in T lymphocytes, such as FLIP, or by upregulation of expressed molecules such as CTLA-4 that are known to alter T lymphocyte sensitivity of membranes to activation induced apoptosis.31,32
Several other antibodies have been reported to increase activation induced T lymphocyte apoptosis. Activation of the human HLA-I α domain caused ceramide-caspase mediated Fas independent apoptosis, whereas horse antilymphocyte serum at a low concentration stimulates apoptosis of phytohaemagglutinin stimulated T lymphocytes through induction of Fas.33,34 Methotrexate also induced Fas independent apoptosis of phytohaemagglutinin stimulated but not resting peripheral blood lymphocytes, and ex vivo activation of peripheral blood lymphocytes of methotrexate treated rheumatoid arthritis patients resulted in apoptosis.35 It has been reported recently that both etanercept, a TNF-α neutralising molecule, and infliximab are strongly synergistic with methotrexate in the treatment of patients with rheumatoid arthritis.36 Although it is not known whether etanercept, like infliximab, alters the susceptibility of T lymphocytes to activation induced apoptosis, it is tempting to speculate that combinations of drugs that alter T lymphocyte apoptosis through different mechanisms may become effective strategies for the treatment of diseases that are associated with local T lymphocyte activation, such as rheumatoid arthritis, multiple sclerosis, and Crohn's disease.37 Such combinations may effectively eliminate the steroid refractory T lymphocyte clones that sustain chronic inflammation in these conditions, and thereby have the potential to modify the natural course of these diseases.
Infliximab treatment has been reported previously to (transiently) induce low titre anti-dsDNA antibodies in 3% of treated patients.7 The much higher incidence of anti-dsDNA antibodies in our study (4/10 patients) is presumably a result of earlier and more frequent sampling in the present study. The mechanism of anti-dsDNA antibody induction by infliximab remains uncertain, but in view of our finding that treatment causes rapid apoptosis of a significant number of lamina propria T lymphocytes, it is tempting to speculate that acute exposure to DNA is one of the causes.
In conclusion, infliximab induced activation dependent apoptosis of Jurkat T lymphocytes, possibly altering the Bax/Bcl-2 ratio. In patients with Crohn's disease, infliximab rapidly increased the number of apoptotic T cells in the inflamed mucosa, without affecting peripheral blood mononuclear cells. These data indicate that infliximab functions, in part, as an “immunotoxin” that specifically targets the mucosal T lymphocytes that are involved in the pathogenesis of Crohn's disease.