Article Text

Abstract
Background: Crohn's disease is characterised by a chronic relapsing inflammation of the bowel in which proinflammatory cytokines play an important perpetuating role. Mitogen activated protein kinase p38 (p38 MAPK) has been established as a major regulator of the inflammatory response, especially with regard to production of proinflammatory cytokines, but its role in inflammatory bowel disease is unexplored. In this paper we describe the effects of a specific p38 MAPK inhibitor, SB 203580, in trinitrobenzene sulphonic acid (TNBS) induced colitis in mice.
Results: SB 203580 had a dichotomal effect in TNBS mice. Weight loss of TNBS mice treated with SB 203580 was significantly worse and colon weight on sacrifice was significantly increased in MAPK inhibitor treated TNBS mice (229.2 mg and 289.1 mg, respectively). However, the total number of cells in the caudal lymph node decreased to 188.8×104 cells in SB 203580 treated TNBS mice compared with 334×104 cells in vehicle treated mice. CD3/CD28 double stimulated caudal lymph node cells of SB 203580 treated mice showed decreased interferon γ production but increased tumour necrosis factor α production. The concentration of interleukin 12p70 in colon homogenates was significantly decreased in SB 203580 treated mice whereas concentrations of interleukin 12p40, tumour necrosis factor α, and interleukin 10 were similar in vehicle and SB 203580 treated TNBS mice.
Conclusion: Our results reveal a dichotomy in p38 MAPK action during experimental colitis.
- colitis
- signal transduction
- T lymphocytes
- SB 203580
- MAPK
- MAPK, mitogen activated protein kinase
- MAPKAP, MAP kinase activated protein kinase
- IL, interleukin
- TNF-α, tumour necrosis factor α
- IFN-γ, interferon γ
- TNBS, trinitrobenzene sulphonic acid
- SDS, sodium dodecyl sulphate
- TBS, Tris buffered saline
- CLN, caudal lymph nodes
- ATF-2, activation of transcription factor 2
Statistics from Altmetric.com
- MAPK, mitogen activated protein kinase
- MAPKAP, MAP kinase activated protein kinase
- IL, interleukin
- TNF-α, tumour necrosis factor α
- IFN-γ, interferon γ
- TNBS, trinitrobenzene sulphonic acid
- SDS, sodium dodecyl sulphate
- TBS, Tris buffered saline
- CLN, caudal lymph nodes
- ATF-2, activation of transcription factor 2
Crohn's disease is a complex multifactorial disorder characterised by cytokine driven T lymphocyte dependent relapsing inflammation of the intestinal mucosa.1–7 The mitogen activated protein kinase (MAPK) family of proteins is the principal regulator of gene expression, and critically controls transcription of a number of cytokine genes. p38 MAPK is particularly involved in the inflammatory process, inflammatory stimuli being strong activators of this kinase, and its activation is required for inflammatory gene transcription in vitro.8 The effects of p38 MAPK on inflammation are probably mediated by direct phosphorylation and activation of several transcription factors (for example, activation of transcription factor 2 (ATF-2)) and downstream protein kinases (for example MAP kinase activated protein kinases 2 (MAPKAP2) and 3 (MAPKAP3)). These downstream kinases regulate cell growth, differentiation, and cell death. The p38 MAPK signalling cascade has a pivotal role in the regulation of transcriptional activation of cytokine genes including interleukin (IL)-1β, tumour necrosis factor α (TNF-α), IL-6, and interferon γ (IFN-γ).9–12
In murine T lymphocytes, a p38 MAPK inhibitor inhibited IFN-γ but not IL-4 production12 and overexpression of dominant negative p38 MAPK results in selective impairment of the Th1 response. Conversely, when highly expressed, constitutively activated MKK6 caused increased production of IFN-γ. The p38 MAPK pathway is also important for other T lymphocyte functions. Mice deficient in MKK3, an upstream kinase of p38 MAPK, are defective in the production of IL-12 by antigen presenting cells and as a result their CD4 positive T lymphocytes do not produce IFN-γ.13 In addition, activation of p38 MAPK was suggested to be necessary for T cell proliferation14,15 although this was not confirmed in other reports.12,16 Finally, p38 MAPK seems to be involved in positive and negative selection of T cells in the thymus.17
The functional importance of p38 MAPK activation has been studied in several animal models using a specific inhibitor. SB 203580 is a selective inhibitor of p38 MAPK that inhibits the catalytic activity of p38 MAPK by competitive binding in the ATP pocket18 and has been shown to inhibit p38 MAPK in vivo. Treatment with SB 203580 reduced mortality in a murine model of endotoxin induced shock19 and had therapeutic activity in collagen induced arthritis in mice.19 Several other inhibitors of p38 MAPK are reported to have efficacy in various disease models including, allergic pulmonary disease,20 lipopolysaccharide induced lung inflammation,21 inflammatory angiogenesis,22 rat adjuvant induced arthritis,23 and pancreatitis associated pulmonary injury.24 In these diseases treatment with p38 MAPK inhibitors attenuated both p38 activation and disease severity. Although it has been suggested that p38 MAPK may be involved in the regulation of intestinal inflammation and is important for cytokine production and T lymphocyte activation, the function of p38 MAPK in inflammatory bowel disease is unknown. In this study we explored the effect of the specific p38 MAPK inhibitor SB 203580 in trinitrobenzene sulphonic acid (TNBS) induced colitis, a murine model of Crohn's disease. The results revealed an unexpected dichotomy in the role of this kinase in inflammatory bowel disease.
MATERIALS AND METHODS
Mice and induction of colitis
All experiments were approved by the Animal Studies Ethics Committee of the University of Amsterdam, the Netherlands. BALB/c mice were obtained from Harlan Sprague Dawley Inc (Horst, the Netherlands). Mice were housed under standard conditions, and supplied with drinking water and food (AM-II 10mm; Hope Farms, Woerden, the Netherlands). Experiments were conducted in eight and 10 week old female BALB/c mice. Colitis was induced by rectal administration of two doses (separated by a seven day interval) of 2 mg of 2,4,6-trinitrobenzene sulphonic acid (TNBS; Sigma Chemical Co, St Louis, Missouri, USA) in 40% ethanol (Merck, Darmstadt, Germany) using a vinyl catheter positioned 3 cm from the anus (10 mice per group). During administration, mice were anaesthetised using isoflurane (1-chloro-2, 2,2,-trifluoroethyl-isoflurane-difluoromethyl-ether; Abbott Laboratories Ltd, Queenborough, Kent, UK), and after administration were kept vertical for 60 seconds. Control mice underwent identical procedures but were given physiological salt. All mice were sacrificed nine days following the first TNBS administration (that is, 48 hours following the second TNBS challenge). Mice were treated daily by intraperitoneal injection of 1 μM SB 203580/kg body weight in 1 ml of sterile saline or 0.01% DMSO in 1 ml of sterile saline as a vehicle control.
To study the kinetics of p38 MAPK activity in TNBS colitis, mice administered TNBS were sacrificed 0, 1, 3, 5, 8, and 9 days after induction of colitis. Mice were treated with either vehicle or SB 203580.
Assessment of inflammation
Body weights were recorded daily. Spleens, caudal lymph nodes (CLN), and colons were harvested on sacrifice. The colons were removed through a midline incision and opened longitudinally. After removal of faecal material the wet weight of the distal 6 cm was recorded and used as an index of disease related intestinal wall thickening. Subsequently, the colons were longitudinally divided into two parts, one of which was used for histological assessment.
Histological analysis
The longitudinally divided colons were rolled up, fixed in 4% formalin, and embedded in paraffin for routine histology. Two investigators who were blinded to the treatment allocation of the mice scored the following parameters: (1) percentage of area involved, (2) number of follicle aggregates, (3) oedema, (4) erosion/ulceration, (5) crypt loss, and (6) infiltration of mono- and polymorphonuclear cells. The percentage of area involved and crypt loss was scored on a scale ranging from 0 to 4 as follows: 0, normal; 1, <10%; 2, 10%; 3, 10–50%; and 4, >50%. Erosions were defined as 0 if the epithelium was intact, 1 for involvement of the lamina propria, 2 for ulcerations involving the submucosa, and 3 when ulcerations were transmural. The severity of the other parameters was scored on a scale from 0 to 3 as follows: 0, absent; 1, weak; 2, moderate; and 3, severe. Hence the score ranged from 0 to a maximum of 26 points.
Cell culture and ELISA for cytokines
CLN cell suspensions were prepared using filter cell strainers (Becton/Dickinson Labware, New Jersey, USA). Cells were suspended in RPMI 1640 medium (BioWhittaker-Boehringer, Verviers, Belgium) containing 10% fetal calf serum and the antibiotic ciproxin (10 μg/ml) (Sigma Chemical Co., St Louis, Missouri, USA). Cell suspensions were counted and 1×105 cells were incubated in 200 μl RPMI containing antibiotics and 10% fetal calf serum in triplicate wells. Cells were stimulated by precoating with anti-CD3 antibody (1:30 concentration; 145.2C11 clone) and soluble anti-CD28 antibody (1:1000 concentration; Pharmingen, San Diego, California, USA). Supernatants were removed after 48 hours and IFN-γ and TNF-α (R&D systems, Abingdon, UK) concentrations measured by ELISA.
Colon homogenates
The colon was harvested and homogenates were made with a tissue homogeniser in nine volumes of Greenburger lysis buffer (300 mM NaCl, 15 mM Tris HCl, 2 mM MgCl2, 2 mM Triton (X-100), pepstatin A, leupeptin, aprotinin (all 20 ng/ml), pH 7.4) Tissue was lysed for 30 minutes on ice followed by two centrifugations (10 minutes, 14 000 g, 4°C). Homogenates were stored at −20°C until use. TNF-α and IL-10 (both R&D), and IL-12p70 and IL-12p40 (both Pharmingen) concentrations were measured by ELISA.
p38 MAPK activity assay and western blotting
Colon homogenates were made using a tissue homogeniser in nine volumes of ice cold cell lysis buffer (20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerolphosphate, 1 mM Na3VO4, 1 μg/ml leupeptin, and 1 mM pefabloc (Merck, Darmstadt, Germany)). Samples were sonicated 4×5 seconds on ice and spun at 7000 g for 10 minutes at 4°C. Protein content in the clear supernatant was determined using a bicinchoninic acid protein assay kit (Pierce, Rockford, Illinois, USA), using bovine serum albumin as the standard, and the supernatant was stored at −80°C.
Approximately 250 μg of protein were used to measure p38 MAPK enzymatic activity with the p38 MAPK assay kit purchased from Cell Signalling (Beverly, Massachusetts, USA). A once diluted slurry of agarose hydrazide bound antibodies to phosphorylated (Thr180/Tyr182) p38 MAPK (40 μl) was used to selectively immunoprecipitate active p38 MAPK from the colon cell lysate (in 200 μl of cell lysis buffer) by gently shaking overnight at 4°C. The immunoprecipitate was washed twice with 500 μl of ice cold cell lysis buffer and twice with 500 μl of ice cold kinase buffer (25 mM Tris, pH 7.5, 5 mM β-glycerolphosphate, 2 mM DTT, 0.1 mM Na3VO4, and 10 mM MgCl2) at 4°C. The kinase reactions were carried out in the presence of 200 μM ATP and 2 μg of ATF-2 fusion protein at 30°C for 30 minutes. After the reaction had been terminated by addition of 3× sodium dodecyl sulphate (SDS) sample buffer, the mixture was boiled for five minutes followed by brief centrifugation. ATF-2 phosphorylation was selectively measured by western immunoblotting. Samples were loaded on 10% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membranes (Millipore, Bedford, Massachusetts, USA). Subsequently, membranes were blocked in 5% non-fat dry milk in Tris buffered saline (TBS) supplemented with 0.1% Tween-20 and washed in TBS supplemented with 0.1% Tween-20. Membranes were incubated overnight using specific antibodies against phosphorylated (Thr71) ATF-2 in 5% bovine serum albumin in TBS supplemented with 0.1% Tween-20. After three washes for 10 minutes, secondary antibody incubation was performed for one hour with GAR-PO in a 1:2000 dilution. After enhanced chemiluminescence using Lumilight+ substrate (Boerhinger Manheim, Germany), antibody binding was visualised using a Lumi-imager (Boerhinger Manheim).
A 250 μg sample of the colon cell lysate was suspended in a final volume of 80 μl SDS sample buffer; 25 μl were loaded on a SDS-polyacrylamide gel to measure p38 MAPK phosphorylation using western blotting, as described previously. p38 MAPK phosphorylation was detected using antibodies against phosphorylated p38 MAPK (Cell Signaling).
Antibody binding was quantified using image analysis software (Boerhinger Manheim) and samples were compared with a control sample set on 100 arbitrary units.
Statistical analysis
Values are presented as mean (SEM) per treatment group. Differences between groups were analysed using the non-parametric Mann-Whitney U test. Body weight changes with time were analysed by one way analysis of variance. p<0.05 was considered significant. SPSS statistical software (SPSS inc., Chicago, USA) was used for analyses.
RESULTS
p38 MAPK is activated during TNBS colitis and inhibited by SB 203580
To investigate the kinetics of p38 MAPK phosphorylation and enzymatic activity, colons from mice subjected to TNBS colitis were harvested over a nine day period. In mice subjected to TNBS and treated with vehicle, enhanced p38 MAPK enzymatic activity was observed, most evident five days after induction of colitis compared with basal levels (fig 1A). Strikingly, after this clear activation on day 5, a reduction in p38 MAPK enzymatic activity was observed. These data were confirmed by direct analysis of p38 MAPK phosphorylation status in colon homogenates which paralleled the kinetics of the kinase assay (fig 1B). The function of p38 MAPK was further explored by analysing the effects of SB 203580, a selective p38 inhibitor, in TNBS colitis. Treatment with SB 203580 almost completely prevented p38 MAPK enzymatic activity in colonic cell lysates induced by TNBS administration (fig 1A). Thus SB 203580 effectively inhibited p38 MAPK at the concentrations used (1 μM/kg body weight/day) in these mice. Analysis of p38 MAPK phosphorylation in SB 203580 treated mice showed no inhibition of phosphorylation status (fig 1B). These data confirm previous reports that SB 203580 binds to the ATP binding site thereby preventing enzymatic activity although not preventing phosphorylation of p38 MAPK by its upstream activators.25
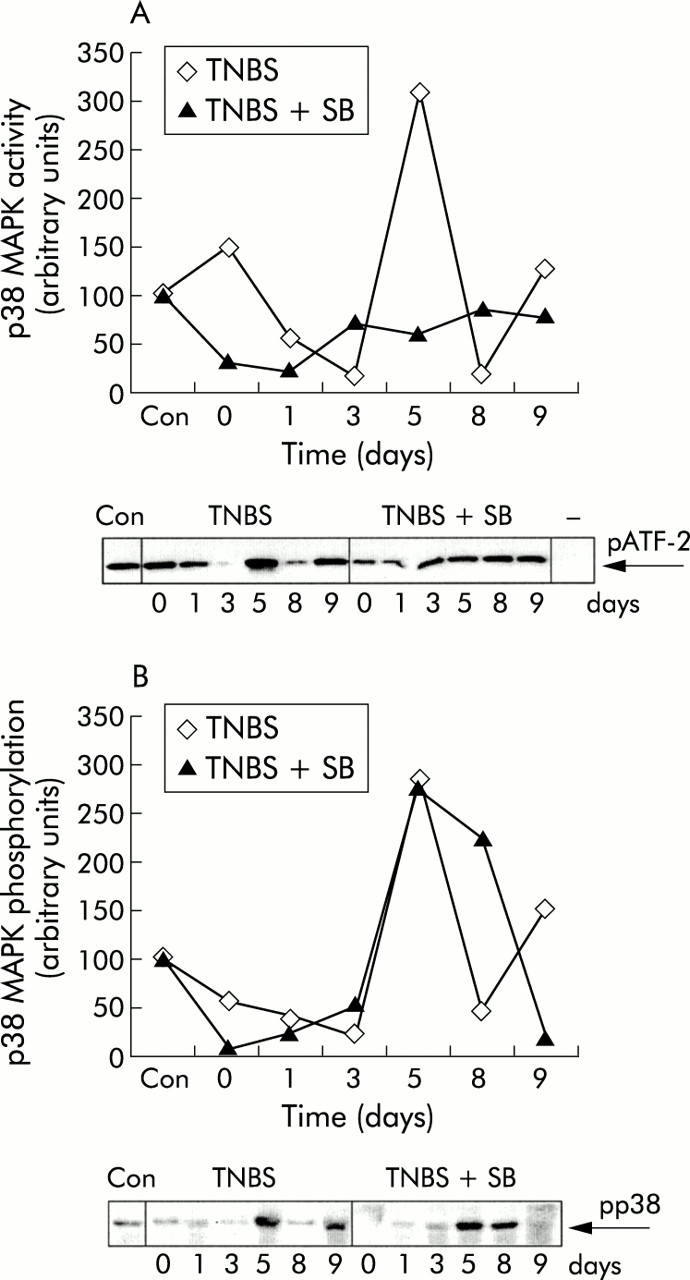
SB 203580 inhibits p38 mitogen activated protein kinase (MAPK) enzymatic activity in vivo. Colonic cell lysates were obtained from trinitrobenzene sulphonic acid (TNBS) instilled mice treated with either vehicle (TNBS) or SB 203580 (TNBS+SB) on days 0, 1, 3, 5, 8, and 9 after the start of the experiment. The colonic cell lysate of an untreated control mouse (Con) was included in both experiments. (A) p38 MAPK enzymatic activity was determined by measuring phosphorylation of activation of transcription factor 2 (pATF-2) in an in vitro kinase assay using immunoprecipitated phosphorylated p38 MAPK from colonic cell lysate. Antibody binding was quantified using image analysis software and expressed compared with a control (Con) that was set on 100 arbitrary units. An immunoprecipitated control is shown (−); only ATF-2 and ATP were mixed, without adding immunoprecipitated p38 MAPK. In SB 203580 treated mice, less enzymatic activity of p38 MAPK was detected. (B) p38 MAPK phosphorylation in colonic cell lysate was determined by western blotting using specific antibodies against phosphorylated p38 MAPK. Antibody binding was quantified using image analysis software, and expressed compared with a control (Con) that was set on 100 arbitrary units.
Mice treated with SB 203580 showed significantly more wasting after induction of colitis
Mice were intrarectally installed with TNBS or saline on day 0 and day 7 and subsequently sacrificed on day 9. All 10 TNBS mice who received vehicle survived but in the TNBS treated mice who received SB 203580 three mice died during the course of the experiment. All mice intrarectally installed with 0.9% NaCl, treated with either vehicle or SB 203580, survived.
Body weights of all mice were recorded daily. Induction of colitis was paralleled by significant weight loss in both groups who received TNBS (fig 2). Mice treated with SB 203580 lost more weight compared with vehicle treated mice with colitis (p<0.05). Vehicle treated mice with colitis started recovering from their initial 15% weight loss on day 3 but in TNBS mice treated with SB 203580 body weight declined further to less then 80% of baseline weight. Mean body weight of TNBS colitis mice treated with SB 203580 remained lower then vehicle treated mice during the course of the experiment. Daily injection of SB 203580 in mice instilled with 0.9% NaCl did not induce weight loss, and body weight changes in these mice were similar to mice treated with vehicle. Hence the effects of SB 203580 on body weight changes during colitis were disease related and did not reflect a generalised effect on p38 MAPK inhibition on murine physiology.

Body weight was recorded daily from day 1 to day 10. The change in weight is expressed as percentage of body weight from day 1 and data are mean (SEM) in saline (NaCl) instilled mice receiving vehicle (n=10), saline instilled mice receiving SB 203580 (n=10), TNBS mice receiving vehicle (n=10), and TNBS mice receiving SB 203580 (n=7).
Colon weights were significantly increased in MAPK inhibitor treated TNBS mice
Colon weight of saline instilled mice treated with vehicle or SB 203580 was similar (81.7 (5.3) mg and 81.3 (6.5) mg respectively) (fig 3). After induction of colitis, colon weight increased to 229.2 (22.2) mg in TNBS colitis, and treatment with SB 203580 caused a further significant increase in colon weight (289.1 (29.1) mg in SB 203580 treated mice) (p<0.05). TNBS administration caused a significant increase in baseline colitis score but on histopathological examination no differences were observed between vehicle and SB 203580 treated TNBS mice. The colitis score of TNBS mice was comparable between vehicle and SB 203580 treated mice (mean total score 13.9 (1.1) and 13.3 (0.4) respectively) (fig 4).

The weight of the last 6 cm of the colon was determined on sacrifice on day 10 after trinitrobenzene sulphonic acid (TNBS) administration. Data are presented as (SEM) in saline (NaCl) instilled mice treated with vehicle or SB 203580 (both n=10) and in TNBS mice treated with vehicle (n=10) or SB 203580 (n=7). *Significant difference.

Haematoxylin-eosin staining of the colon of BALB/c mice treated with SB 203580 or vehicle (magnification 25× object). (A) Colon of saline instilled mouse treated with vehicle, showing normal architecture. (B) Colon of a saline instilled mouse treated with SB 203580, also showing normal architecture. (C) Colon of a trinitrobenzene sulphonic acid (TNBS) instilled mouse treated with vehicle; common histological features include ulceration and influx of cells. (D) Colon of a TNBS instilled mouse treated with SB 203580; common inflammatory features are also seen.
Cellularity of caudal lymph nodes in SB 203580 treated TNBS mice
The total number of cells in the CLN that drain the inflamed colon increased in TNBS colitis from 65.7 (17.1) ×104 cells (saline) to 334 (36.7) ×104 cells (TNBS) (fig 5). Significantly less CLN cells were found in SB 203580 treated mice with colitis compared with vehicle treated mice with colitis (188.6 (44.9) ×104 cells in SB 203580 treated TNBS mice; p<0.05). This effect of SB 203580 in control mice was not observed in saline instilled mice (52.1 (11.2) ×104 cells). Apparently, p38 MAPK is an important regulator of CLN cell number.

Total number of cells present in the caudal lymph node on sacrifice. Data are presented as mean (SEM) in saline (NaCl) instilled mice treated with vehicle or SB 203580 (both n=10) and in trinitrobenzene sulphonic acid (TNBS) mice treated with vehicle (n=10) or SB 203580 (n=7). *Significant difference.
Cytokine production
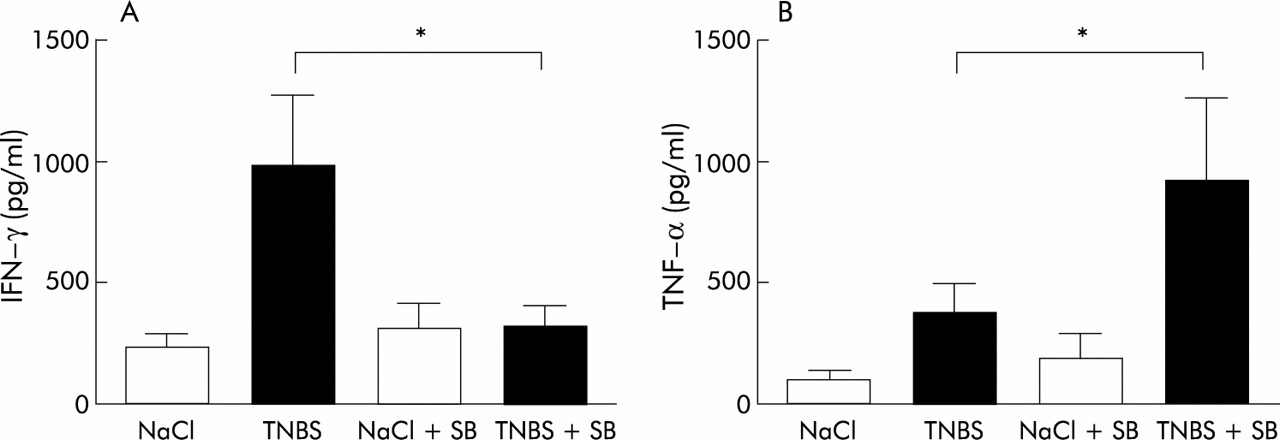
Cytokine production on activation of T cells in the CLN was determined by CD3/CD28 double stimulation (fig 6). In TNBS colitis, IFN-γ production of CLN cells increased and SB 203580 treatment reduced IFN-γ production from 981.3 (287.8) pg/ml (in vehicle treated TNBS mice) to 305.3 (83.01) pg/ml (fig 6A). In saline instilled mice, IFN-γ production of CLN cells was similar in vehicle treated and SB 203580 treated mice (228.9 (60.8) pg/ml and 293.8 (116.7) pg/ml, respectively). TNF-α production of stimulated CLN cells was increased in TNBS mice (fig 6B). Unexpectedly, treatment of TNBS mice with SB 203580 resulted in higher TNF-α production compared with vehicle treated TNBS mice (375.3 (125.9) pg/ml in TNBS mice and 921.58 (351.2) pg/ml in TNBS mice treated with SB 203580). In saline instilled mice, treatment with SB 203580 only slightly increased TNF-α production by CLN cells (188.4 (100.6) pg/ml in SB 203580 treated mice and 108.3 (37) pg/ml in vehicle treated control mice).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cytokine production by stimulated caudal lymph node cells. Caudal lymph node cells were stimulated with CD3/CD28, and interferon γ (IFN-γ) (A) and tumour necrosis factor α (TNF-α) (B) production was measured after 48 hours. Data are presented as mean (SEM) in saline (NaCl) instilled mice treated with vehicle or SB 203580 (both n=10) and in trinitrobenzene sulphonic acid (TNBS) mice treated with vehicle (n=10) or SB 203580 (n=7). *Significant difference.
In colon homogenates, IL-12p70 and IL-12p40 concentrations were measured by ELISA (table 1). A significant reduction in IL-12p70 (p40/p35 heterodimer) concentration was observed in TNBS mice treated with SB 203580 (133.5 (8.5) pg/mg and 91.2 (11.9) pg/mg, respectively.). No difference in total IL-12p40 concentration was detected in the colon homogenates of TNBS mice (mean 1492 (112.8) pg/mg and 1483 (124.7) pg/mg, respectively; p<0.05). TNF-α concentrations in colon homogenates were similar in TNBS mice treated with SB 203580 or vehicle (179.6 (6.5) pg/mg and 188.1 (8.5) pg/mg, respectively.). In addition, no changes in IL-10 concentrations were detected in colon homogenates of TNBS mice treated either with vehicle or SB203580 (470.9 (64.7) pg/mg and 462.0 (42.8) pg/mg, respectively).
Cytokine concentrations in colon homogenates measured by ELISA
DISCUSSION
Our study was designed to explore the role of p38 MAPK in inflammatory bowel disease. To this end, we pharmacologically inhibited p38 MAPK during TNBS colitis using the specific p38 MAPK inhibitor SB 203580. SB 203580 was previously shown to be a highly selective inhibitor of p38 MAPK and at a concentration of 1 μM did not affect a wide range of other kinases, including p42 and p54 MAPK, and phosphatases.10,26 In agreement with the reported p38 MAPK inhibitory activity of SB 203580, we showed a significant reduction in p38 MAPK activity in colonic cell lysates in mice treated with SB 203580. Furthermore, we found transient activation of p38 MAPK in TNBS instilled mice. Both phosphorylation and enzymatic activity were at a maximum five days after the start of the experiment.
Surprisingly, inhibition of p38 MAPK with SB 203580 in mice with TNBS induced colitis showed dual effects. Mice treated with SB 203580 lost more weight and had higher colon weights. However, we found a reduction in the number of cells present in the CLN. It should be noted that this antiproliferative response could be explained by a specific action of SB 203580 rather than p38 inhibition. Several reports describe inhibition of T cell proliferation using the inhibitor SB 20358010,14,27 but mice with dominant negative p38 and T cells treated with a different p38 MAPK inhibitor,12 CNI-1493, showed normal T cell proliferation.28 In contrast, the reduction in cell number may be explained by induction of apoptosis in the CLN of mice treated with SB 203580. However, SB 203580 was reported to specifically inhibit induction of T cell apoptosis.27,29 Therefore, is seems that the reduction in cell number could be better explained by diminished proliferation.
In line with previous reports, stimulated CLN cells produced significantly less IFN-γ.12,27,30,31 Furthermore, treatment resulted in significantly lower IL-12p70 concentrations in the colon whereas IL-12p40, TNF-α, and IL-10 concentrations in the colon were unaffected. Interestingly, TNF-α production of CLN cells was increased by SB 203580 treatment. Thus despite reduction of IFN-γ and IL-12p70 production, treatment with SB 203580 exacerbated the disease.
There are several explanations for this unexpected observation. We have recently reported that IFN-γ is not an important disease mediator in TNBS colitis because mice that lack IFN-γ32 or its receptor33 are susceptible to TNBS colitis and, in fact, develop more severe disease. It was reported that IL-12 synthesis and specifically IL-12p40 synthesis are dependent on p38 MAPK activation.13 We found a specific reduction in IL-12p70 and no changes in IL-12p40 concentrations after SB 203580 treatment. The pathogenic importance of IL-12 in TNBS colitis is well known and IL-12 neutralising antibody treatment is highly protective in this model.34 It should be noted that the two IL-12 subunits, p35 and p40, have different biological functions. IL-12p35 deficient mice are protected against the development of TNBS induced colitis, and in this situation neutralisation of p40 restores the normal sensitivity to TNBS.35 Hence IL-12p40 is protective, possibly through the activity of IL-12 homodimers or dimerisation with another ligand. Against this background, the combination of a reduction in IL-12p70 without an effect on IL-12 p40 concentration would be expected to be protective but this was not substantiated in our study. The observed changes may have been too small to result in a therapeutic effect or IL-12p35 may have formed heterodimers with a ligand other than p40. It is known that heterodimers of IL-12p35 and Epstein-Barr virus induced gene 336 have proinflammatory effects.
The enhanced wasting observed during colitis in p38 MAPK inhibited animals coincided with enhanced TNF-α production. In accordance with this finding, TNF-α production of T lymphocytes was reported to be only partially dependent on p38 MAPK activation.37 Furthermore, we have recently shown that SB 203580 increases TNF-α production by macrophages38 and increased TNF-α production in mast cells has been reported.39 TNF-α plays an important role in intestinal inflammation, as confirmed in Crohn's disease1,40 and experimental colitis.41
Alternatively, it is possible that activation of p38 MAPK occurs downstream of the disease perpetuating signal transduction elements and that inhibition at this level does not affect disease severity. We consider it more likely that cells that play a protective role in intestinal inflammation also need p38 MAPK to function properly. T regulatory cells that produce large amounts of IL-10 are protective in experimental colitis in mice.42 Although we did not find a reduction in IL-10 concentration in colon homogenates of mice treated with SB 203580, it was reported that SB 203580 suppresses IL-10 production of T cells31 and hence SB 203580 could inhibit the protective T cells present in the lamina propria.
Another explanation for our findings is that inhibition of p38 MAPK results in accumulation of upstream activators of p38 MAPK. SB 203580 binds to the ATP binding site thus preventing phosphorylation of downstream targets, including MAPKAP 2 and ATF-2, although not preventing phosphorylation of p38 MAPK by its upstream activators MKK3 and MKK6.25 MKK3 and MKK6 can phosphorylate downstream targets other than p38 MAPK, such as the JNK1 and JNK2 pathway.43 Because JNK2 is involved in Th1 differentiation, p38 MAPK inhibition may paradoxically increase the severity of Th1 mediated diseases. It is however also possible that the observed effects are specific for the inhibitor or species used in these experiments and studies with other p38 MAPK inhibitors are needed to elucidate this.
In summary, treatment with the p38 MAPK inhibitor SB 203580 does not ameliorate TNBS colitis although it does prevent IFN-γ and IL-12p70 production. This indicates that p38 MAPK may have a broader role in the mucosal immune response and is not only responsible for the production of proinflammatory cytokines but may also be involved in counterregulatory responses.