Article Text

Abstract
Background: Non-cirrhotic portal hypertension of unknown cause is a poorly understood condition attributed to obstructive portal venopathy.
Aim: To reassess the manifestations, course, and causes, with special attention to thrombosis.
Methods: Analysis of a cohort of 28 patients.
Results: Gastrointestinal bleeding occurred in 11 patients. Liver failure developed at the time of concurrent disease in eight patients, including all four patients who died. Portal vein thrombosis developed in 13 patients. A prothrombotic disorder was found in 12 of 23 fully investigated patients. Hepatoportal sclerosis was observed in 11 patients (with associated perisinusoidal fibrosis and/or nodular regenerative hyperplasia in six); periportal fibrosis, perisinusoidal fibrosis, nodular regenerative hyperplasia, or a combination thereof were observed in other patients. A morphometric evaluation showed an increased number of portal vessels in patients with hepatoportal sclerosis. There was no relation between pathological results and haemodynamic findings or prothrombotic disorders.
Conclusions: Outcome was related to associated conditions. Overlap in pathological, haemodynamic, and causal features suggests a single entity, with prothrombotic disorders as major causal factors, and injury to sinusoids as well as to portal venules as the primary mechanism. Activated coagulation could mediate vascular injury in the absence of thrombosis. Anticoagulation should be considered.
- portal hypertension
- hepatoportal sclerosis
- perisinusoidal fibrosis
- prothrombotic disorder
- portal vein thrombosis
- liver failure
- portal venules
- NCIPH, non-cirrhotic intrahepatic portal hypertension
- NASH, non-alcoholic steatohepatitis
- HBsAg, hepatitis B surface antigen
- anti-HBs, antibodies against hepatitis B surface antigen
- anti-HBc, antibodies against hepatitis B core antigen
Statistics from Altmetric.com
- portal hypertension
- hepatoportal sclerosis
- perisinusoidal fibrosis
- prothrombotic disorder
- portal vein thrombosis
- liver failure
- portal venules
- NCIPH, non-cirrhotic intrahepatic portal hypertension
- NASH, non-alcoholic steatohepatitis
- HBsAg, hepatitis B surface antigen
- anti-HBs, antibodies against hepatitis B surface antigen
- anti-HBc, antibodies against hepatitis B core antigen
Non-cirrhotic intrahepatic portal hypertension (NCIPH) is characterised by an increased portal pressure with patent portal and hepatic veins, in the absence of cirrhosis.1,2 The causes of NCIPH are varied.1,2 Schistosomiasis is the most common cause of NCIPH. Alcoholic, metabolic, or autoimmune liver diseases can cause portal hypertension at the precirrhotic stage. Various substances causing NCIPH have been identified, some being taken for therapeutic or diagnostic purposes. In many cases however the cause remains unknown and NCIPH is then referred to as idiopathic. Idiopathic NCIPH has been reported mainly in India and Japan.3–,9
Various terms have been given to this entity: idiopathic portal hypertension,9 non-cirrhotic portal fibrosis with portal hypertension,6 benign intrahepatic portal hypertension,7 hepatoportal sclerosis,10 nodular regenerative hyperplasia,8 and incomplete septal cirrhosis.11 Many aspects of idiopathic NCIPH are unclear. For example, the clinical manifestations are related to portal hypertension, and liver function seems to be preserved so that evolution has been regarded as benign.1,2,7 Recently however, liver failure has been reported.12 Pathologically, two types of lesions have been described: alterations in small vessels, regarded as the initial lesion,8,13 and changes in liver architecture consisting of fibrosis and/or nodule formation, regarded as secondary.8 However, obstructive portal venopathy, the suspected initial lesion, is not always found in the biopsy specimen. Furthermore, the mechanisms causing these lesions remain largely conjectural. In particular, the role of thrombosis and prothrombotic disorders in causing obstructive portal venopathy has not been investigated.
The aim of this study in a recent cohort of Western patients with idiopathic NCIPH was to re-evaluate the clinical manifestations, haemodynamics, course, pathological features, and causes, with particular attention to thrombosis and prothrombotic disorders.
PATIENTS AND METHODS
Case definition and patient selection
The diagnosis of idiopathic NCIPH was considered when the following criteria were fulfilled: (i) evidence of portal hypertension (any one of the following features: oesophageal varices, hypersplenism, ascites, or increased hepatic venous pressure gradient); (ii) Doppler ultrasound showing patent portal and hepatic veins at the time of diagnosis of NCIPH; (iii) liver biopsy showing no cirrhosis; (iv) exclusion of conditions causing cirrhosis according to conventional diagnostic criteria (chronic viral hepatitis, alcoholic liver disease, non-alcoholic steatohepatitis (NASH), obesity, haemochromatosis, autoimmune hepatitis, or Wilson's disease)1,2; and (v) exclusion of chronic vitamin A intake, professional exposure to copper sulphate, vinyl chloride monomer, past angiography with thorium sulphate, exposure to Spanish toxic oil, or arsenic salts. We excluded these risk factor by repeated interview with the patient, his or her relatives, and the attending physicians, and by chemical determinations of vitamin A and arsenic in biological material, as appropriate.1,2 We considered evidence of NASH the association of steatosis, ballooning degeneration of hepatocytes, and lobular, mixed inflammatory infiltrates. All patients with nuclear, smooth muscle, liver kidney microsomes, or mitochondrial autoantibodies in serum were considered as potentially affected with an autoimmune chronic liver disease and therefore excluded.
Haemodynamics
Cannulation of the hepatic veins for measurement of free and wedged hepatic venous pressures was performed according to previously reported methods.14 Only patients in whom there was an indication for transvenous liver biopsy underwent a haemodynamic study.
Histological evaluation
Microscopic evaluation was performed by two pathologists (EB and CD). Liver sections were stained with haematoxylin-eosin, Masson's trichrome, and reticulin argentation (Gomeri's method). Four pathological entities were considered: (i) hepatoportal sclerosis when intimal fibrous thickening of the portal vein or aberrant portal vessel was evident10; (ii) periportal fibrosis (fibrous enlargement of the portal tracts with or without extending fibrous septa); (iii) perisinusoidal fibrosis as shown by the reticulin argentation; and (iv) nodular regenerative hyperplasia when a nodular architecture without extensive fibrosis was demonstrated or when there was an association of thickened liver cell plates alternating with atrophic compressed ones, according to the description by Wanless.8 Using a CD34 monoclonal antibody (clone QBEND 10; Immunotech, Marseille, France; dilution 1/50) as an endothelial marker,15 we determined the mean number of portal vascular lumens per portal tract. At least three portal tracts were examined at a magnification of 400 (mean 5.6 portal tracts examined, range 3–11). Protocol surgical specimens from 10 normal liver allografts, taken following revascularisation at the end of liver transplantation, were used as controls.
Investigations for a prothrombotic state
Endogenous erythroid colonies and total red cell mass were assessed as previously described.16,17 Overt polycythemia vera was diagnosed by a haematocrit >125% of calculated normal values, and suggestive bone marrow findings18; the so-called forme fruste of the primary myeloproliferative disorder was characterised by endogenous erythroid colonies, by normal or decreased total red cell volume with normal iron stores, and by the absence of other well characterised myeloproliferative disorders.19 Evaluation of the CD55-CD59 cluster was performed to exclude paroxysmal nocturnal haemoglobinuria.20 Antithrombin and protein C functional deficiencies, and total and/or free protein S antigenic deficiencies were assessed using commercial kits (Diagnostica Stago, Asniéres, France) and only considered when isolated—that is, when levels of other inhibitors or clotting factors were normal so that hepatocellular insufficiency could be ruled out. A commercial kit was used to assay antiphospholipid antibodies of IgG and IgM types (Biogenic, Montpellier, France). Lupus anticoagulant was investigated as previously described.21 Genetic analyses were performed according to previously reported methods for G1691A factor V22 and G20210A prothrombin.23
RESULTS
From January 1994 to September 1998, lesions consistent with NCIPH were found at pathological examination in 42 patients in the absence of any of the known causes of chronic liver disease or NCIPH. Fourteen patients were excluded: three did not have sufficient evidence of portal hypertension and 11 had thrombosis of the portal vein documented at the time of diagnosis of NCIPH. Histological lesions and prothrombotic disorders of 10 of these 11 patients with both histopathological features of NCIPH and portal vein thrombosis at the time of diagnosis are presented in table 1⇓. The remaining 28 patients formed the basis of this study.
Histopathological findings and prothrombotic disorders in patients with both liver lesions consistent with non-cirrhotic intrahepatic portal hypertension (NCIPH) and portal vein thrombosis at the time of diagnosis of NCIPH
Demographics, manifestations, and haemodynamics
There were 20 men and eight women, aged 41.8 (15) years at the time of initial presentation. Initial manifestations were related to portal hypertension in 22 patients (variceal bleeding in nine patients, non-bleeding varices in three, splenomegaly in 10). Abnormal results of liver function tests and other miscellaneous manifestations accounted for the remaining two and four patients, respectively. Results of liver function tests at presentation are presented in table 2⇓. Mild alterations were usual. However, in eight patients major alterations in the results of liver function tests were found at initial presentation. Measurement of hepatic venous pressures was performed in 13 patients. Individual results are shown in table 3⇓. Hepatic venous pressure gradient was normal (<5 mm Hg) in five patients and increased (median 9 mm Hg (range 6–21)) in the remaining eight patients.
Results of liver function tests at initial presentation and at the end of follow up
Pathological findings, prothrombotic states, and features of portal hypertension
Pathological features
Liver specimens were obtained by needle biopsy in 25 patients (through the transcapsular route in 15, through the transvenous route in 13), at surgery in six patients, and at necropsy in one patient. Nine patients had repeated liver sampling. In seven of these nine patients, repeated sampling was needed because the initial biopsy did not permit pathological diagnosis. Individual results of pathological examinations are presented in table 3⇑. Of the 28 patients, 11 had hepatoportal sclerosis (see fig 1⇓). Among these 11 patients, six had associated lesions: three had perisinusoidal fibrosis and three had nodular regenerative hyperplasia. Among the 17 patients without hepatoportal sclerosis, nine had periportal fibrosis, seven perisinusoidal fibrosis, and two nodular regenerative hyperplasia. Two patients had associated periportal and perisinusoidal fibrosis. One remaining patient whose slides could not be reviewed by us was considered unclassified (patient No 28).

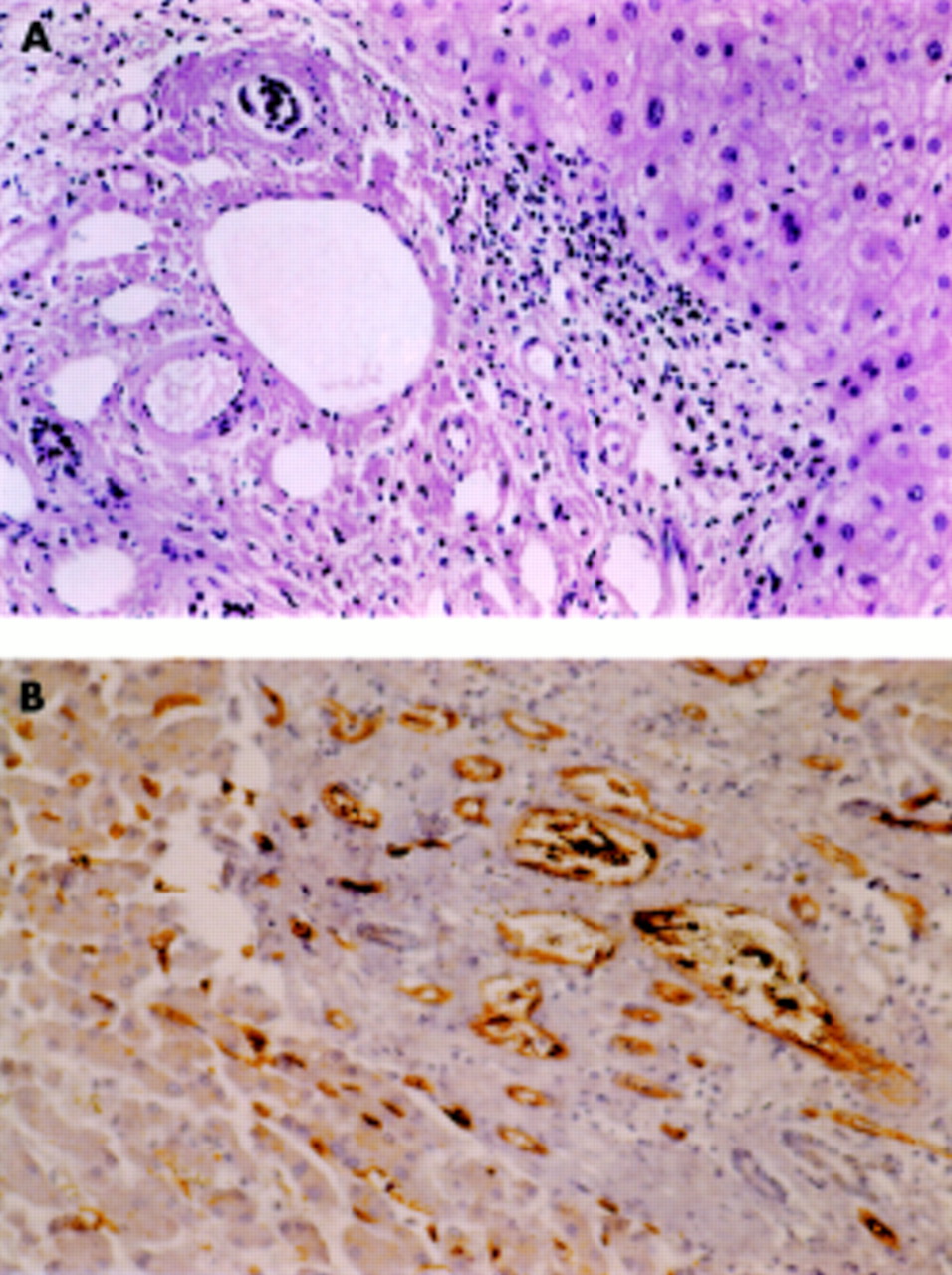{kind=link}
Hepatoportal sclerosis. (A) Haematoxylin and eosin stain showing an increased number of vascular channels. (B) Immunostaining with anti-CD34 demonstrating endothelial lining in multiple small channels within another portal space in the same patient.
Among the 28 patients, 16 had at least one liver specimen in which the mean number of portal vessels could be evaluated semiquantitatively; eight belonged to the hepatoportal sclerosis category (with nodular regenerative hyperplasia in two) and eight to the periportal or perisinusoidal fibrosis category. A mean of 5.6 portal tract were examined (range 3–11). The number of vascular sections per portal tract observed in normal control livers was 3.3 (range 1–6). The mean number of vascular sections per portal tract was higher when hepatoportal sclerosis was present (11.1 (range 5.6–23.8)) than when it was absent (3.4 (range 2.3–5.6)) (p<0.05). In four patients, evaluation of vascular sections repeated in a second specimen gave similar results (table 1⇑, patient Nos 2, 21, 23, 24). There was no difference in hepatic venous pressure gradient according to the four main types of pathological findings.
Prothrombotic disorders
Six patients had a familial history of thrombosis. Two were found to have an inherited prothrombotic disorder. Extensive investigation for prothrombotic disorders was carried out in 23 of 28 patients but was incomplete in the other five patients. A prothrombotic disorder was found in 12 (48%) patients. Myeloproliferative disorder was shown in five patients (overt polycythemia vera in one and forme fruste in four). Of these five patients, two also had protein S deficiency. Protein S deficiency was found in two additional patients and protein C deficiency in one other. Elevated titres of anticardiolipin antibodies were found in five patients (one with another prothrombotic state and four without). The distribution of prothrombotic states according to the four types of pathological findings is presented in table 2⇑. Every type of prothrombotic disorder (myeloproliferative disorder, antiphospholipid syndrome, or deficiency in natural anticoagulant protein) was encountered in every pathological category.
Outcome
Mean duration of follow up after the first manifestations was 7.6 years (range 1–21). Ultrasound or computed tomography scans were repeated in all patients. Portal vein thrombosis developed in 13 patients. Nine of these 13 patients had a local precipitating factor or an associated prothrombotic state: primary myeloproliferative disorder in two patients; protein C deficiency and primary myeloproliferative disorder in one; splenectomy, protein S deficiency, and primary myeloproliferative disorder in one; sclerotherapy in two, including one who also had abdominal sepsis; surgical portocaval shunt in one; adenocarcinoma of the colon in one; and splenectomy and protein S deficiency in one. One or several episodes of gastrointestinal bleeding occurred in six and 11 patients, respectively. Prevention of recurrent bleeding was carried out using beta adrenergic blocking agents at initial therapy, and endoscopic therapy when there was recurrent bleeding on beta adrenergic blocking agents. Surgical portocaval shunts were constructed in three patients because of recurrent bleeding, despite endoscopic therapy in two and, in the remaining patient, at the time of surgery for an abdominal abscess complicating endoscopic therapy in the absence of recurrent bleeding. None of these patients treated with a shunt developed encephalopathy over a two year follow up period. Of the 23 patients receiving beta adrenergic blocking agents for prevention of first or recurrent bleeding, five bled or rebled while on therapy. Ascites developed in 14 patients; 11 had gastrointestinal bleeding or severe concurrent disease (infectious disease in three, abdominal surgery for adenocarcinoma of the colon in two, acute B viral hepatitis in one, chronic renal failure in three, and type I diabetes in two). Ascites was transient in 10 patients and chronic in four. Transient ascites was associated with gastrointestinal bleeding, concurrent infection, or surgery, while chronic ascites was associated with insulin dependent diabetes (n=2) or renal failure (n=3). Manifestations of liver failure developed in six patients. Again, these manifestations followed gastrointestinal bleeding or abdominal surgery or were associated with severe concurrent condition. As a rule, liver function tests improved when the intercurrent conditions or complications were controlled. However, as showed in table 2⇑, the results were similar, on average, at presentation and at the end of follow up. Only two patients had repeated biopsy during follow up specimens, allowing for comparison of the pathological features. There were no obvious changes. None of these two patients had developed cirrhosis.
Four patients died, all of whom had terminal liver failure. Of these four patients, one patient had superimposed fulminant viral B hepatitis occurring 12 years after the diagnosis of NCIPH (the autopsied patient); he tested negative for hepatitis B surface antigen (HBsAg), antibodies against hepatitis B surface antigen (anti-HBs), and antibodies against hepatitis B core antigen (anti-HBc) in serum prior to the acute episode whereas he tested positive for HBsAg and anti-HBc-IgM in serum at the time of acute hepatitis. Two additional patients had terminal chronic renal failure that was attributed to their severe insulin dependent diabetes; one had terminal chronic renal failure of uncertain cause and metastatic prostatic cancer. Thus a severe associated condition contraindicated liver transplantation in these four patients. In two other patients, liver transplantation was considered but their condition improved with control of the intercurrent infectious disorder (septic portal thrombosis following endoscopic therapy of oesophageal varices in one patient and Epstein-Barr virus infection in another).
Portal vein thrombosis was documented in three of four patients who died and in 10 of 24 patients who survived. Three of 13 patients with portal vein thrombosis died while one of 15 patients without portal vein thrombosis died (p=0.2).
DISCUSSION
The largest surveys of idiopathic NCIPH were reported more than 15 years ago and included patients seen more than 15 years earlier.3–5,24 Their conclusions needed to be reassessed because at that time current markers for viral hepatitis and other chronic liver disorders were not available and non-surgical therapy for portal hypertension was not widely used. A cohort of 42 patients with incomplete septal cirrhosis—a particular form of NCIPH11—was recently reported from Belgium25 but patients with a known cause of chronic liver disease or a known cause of NCIPH such as arsenic intoxication had not been excluded. The present cohort consisted of 28 Western patients with idiopathic NCIPH without initial extrahepatic portal vein thrombosis, and were followed up for several years. Our study suffered from some of the limitations of retrospective surveys but these were kept to a minimum as the whole cohort was enrolled and the end points were chosen to be simple and objective. Central to this study was exclusion of cirrhosis, based not only on pathological examination of biopsy specimens but also on exclusion of all potential causes of cirrhosis. Necropsy specimens suffer from the drawback of sampling only the advanced stage of the disease, and surgical wedge biopsy from being unrepresentative of the deeper part of the liver.1–,3
Similar to the findings in India and Japan, portal hypertension accounted for 70% of the initial manifestations, and signs of mild liver function impairment were observed in the majority of patients.3–,11 Decompensated liver disease or liver failure developed in 33% of patients. It is worth noting however that these changes either followed severe gastrointestinal bleeding or were associated with severe concurrent disease. When these associated conditions were controlled, liver function recovered. It is tempting to explain impaired liver function by incipient atrophy of hepatic parenchyma due to ischaemia, as suggested by angiographic studies.3 However, other reasons for alterations in the results of liver tests should be considered: decreased coagulation factor levels could be explained by activation of coagulation26 or hypersplenism27; increased serum bilirubin by an associated sepsis, or by destruction of transfused erythrocytes; and decreased serum albumin by dilution due to bleeding and volume replacement therapy. In two patients in the present cohort, interpretation of marked but transient abnormalities of liver test results as evidence of irreversible liver failure would have led to unnecessary transplantation.
Until recently, NCIPH was considered a form of portal hypertension with a good prognosis compared with cirrhotic portal hypertension.1–11,25 Our findings agree with this view as none of our patients died as a result of complications of portal hypertension. Prophylaxis for gastrointestinal bleeding in our patients was based on beta blocking agents and endoscopic therapy. However, the number of patients was too small to allow conclusions regarding efficacy. Contrasting with the view of a benign course however, liver failure requiring liver transplantation has been described recently.12 Indeed, in our cohort, four patients died, all of whom had terminal liver failure. However, all four patients had a severe associated condition which both contributed to the downhill course and contraindicated liver transplantation. In three of the four patients who died, terminal renal failure was present, suggesting that the metabolic consequences of renal failure interacted in some way with liver atrophy to produce liver failure. Therefore, our findings confirm that isolated idiopathic NCIPH is a relatively benign disorder but also indicate that idiopathic NCIPH makes the patient more sensitive to the deleterious effects of concurrent conditions. The course of liver lesions remains unknown because in this study systematic follow up liver biopsy was not considered justified for the management of patients, and the only patient in whom necropsy could be obtained died from superimposed fulminant viral hepatitis.
There was a trend towards portal vein thrombosis being associated with a poor prognosis. It will be interesting to address this issue in a larger cohort of patients. Superimposed thrombosis of the portal vein is a poor prognostic factor in Budd-Chiari syndrome.28 Experimental portal vein obstruction induces liver cell loss, the degree of which is related to the degree of portal venous obstruction.29 Therefore, maintenance of portal vein patency may become an important goal for therapy in NCIPH.
NCIPH has been associated with a variety of lesions, including abnormal portal vessels, periportal or septal fibrosis, perisinusoidal fibrosis, sinusoidal dilatation, and nodule formation.30,31 The present findings confirm this heterogeneity and indicate a number of overlapping features between several pathological entities: hepatoportal sclerosis, periportal fibrosis, perisinusoidal fibrosis, and nodular regenerative hyperplasia. We were unable to disclose any specific association between these pathological entities and clinical, haemodynamic, causal, or prognostic features. In particular, the lack of concordance between the lesion of hepatoportal sclerosis and haemodynamic evidence of a presinusoidal block suggests that some form of sinusoidal block is present, for example in the form of perisinusoidal fibrosis inconspicuous in the liver sample examined but possibly more marked in other parts of the liver. Moreover, electron microscopy may have revealed perisinusoidal fibrosis that was not visible even using the reticulin stain.32 Thus apparently idiopathic NCIPH can be viewed as a single entity with various pathological aspects rather than separate entities. Based on necropsy findings, a unifying theory of obstructive portal vasculopathy has been put forward in which alterations in liver architecture and deposition of fibrous tissue follow the initial injury to the portal venules.8 Some of the present findings indirectly support this view. Specifically, in the category of hepatoportal sclerosis, we have demonstrated by means of morphometry an increased number of portal vascular channels which can be regarded as equivalent to intrahepatic cavernoma bypassing portions of obstructed portal venules.13,33 An alternative but less solid interpretation would be that these aberrant vessels are dilated lymphatics related to increased lymph flow because of portal hypertension.34
Other findings however are at variance with the obstructive portal vasculopathy theory. Abnormal portal vessels were found in less than half of the cases. Morphometric evaluation of portal vascular sections with a sensitive endothelial marker did not increase this proportion. Nodular regenerative hyperplasia was associated with conspicuous alterations of portal vessels in only three of five cases. Periportal and perisinusoidal fibrosis were more commonly encountered in the absence than in the presence of portal vessel alterations. A simple explanation could be insufficient sampling through needle aspiration or surgery compared with necropsy specimens on which the obstructive portal venopathy theory was elaborated.8,31,35,36 However, it can be argued that necropsy specimens do not account for early lesions. Furthermore, a cause and effect relationship between obstructive portal venopathy and architectural alterations should be visible in consecutive sections of the same specimen. Therefore, our findings suggest an alternative hypothesis in which the causative process acts directly on the sinusoidal wall as well as on the portal vein wall to induce fibrosis, obstruction, and secondary alterations in the architecture, as previously suggested by others.5,6,37,38 Stellate cell activation is induced by thrombin.39 Stellate cell activation by various endogenous factors, including cytokines and activated coagulation factors, might prove relevant to the formation of perisinusoidal fibrosis in idiopathic NCIPH in the absence of clear portal venular obstruction.
The mechanism of vascular alterations in NCIPH has not been explained. We hypothesised that a prothrombotic disorder may be present and cause obstruction of small vessels just as occurs in large vessels.16,17,36–,42 The prevalence of identified prothrombotic disorders in the present cohort (12 (54%) of 23 fully investigated patients) was higher than that expected in the general Western population (8–15%),43 which argues for the role of coagulation activation in NCIPH. Similar findings have been presented by other investigators in a preliminary form.42 However, the importance of unidentified factors cannot be ruled out. The rarity of NCIPH and the wide variety of currently recognised causal factors indicate that NCIPH is likely to be a multicausal disease, with prothrombotic disorders as one of the causal factors.
The idea that prothrombotic disorders play a role in NCIPH is reinforced by our finding that 13 of 28 patients developed extrahepatic portal vein thrombosis during follow up and that nine of these 13 patients had a prothrombotic state or a local factor. Furthermore, 11 patients with portal vein thrombosis at the time of histopathological diagnosis of NCIPH had a spectrum of liver lesions and a frequency of associated prothrombotic conditions similar to those of patients without portal vein thrombosis at the time of diagnosis. However, contrasting with the frequency of extrahepatic portal vein thrombosis, recent thrombi were almost never seen in the small sized portal veins in this and other pathological studies; rather, fibrous wall thickening of the medium and large sized intrahepatic veins has usually been reported4,6,10,17,30,44,45 and sometimes interpreted as old recanalised thrombi, although a process similar to that of arterial sclerosis was also considered.
In summary, in Europe, as in the Far East, the usual manifestations of NCIPH are those of portal hypertension. Features of liver failure occur when there is an associated severe condition. Prognosis is related to the associated conditions. Pathological findings include, alone or in combination, hepatoportal sclerosis, periportal and perisinusoidal fibrosis, and nodular regenerative hyperplasia. Aberrant vessels, equivalent to a microscopic intrahepatic portal cavernoma, can be found in one third of patients. The primary injury may be to the portal venules or to the sinusoids. Prothrombotic disorders are found in 50% of patients. Activation of coagulation is likely to play a central role. Extrahepatic portal vein thrombosis occurs in half of the patients. Therefore, anticoagulant therapy should be strongly considered.
Acknowledgments
Financial support was obtained from Fonds d'études et de recherches du corps médical des hôpitaux de Paris. The authors are indebted to Dr Alain Cazier (Service d'Anatomie et de Cytologie Pathologiques, Centre Hospitalier Laennec, 60109 Creil, France) for his collaboration.