Article Text

Abstract
Background and aims: Mutant tumour derived DNA has been detected in the sera of colorectal cancer patients. We investigated if mutant serum KRAS2 was detectable preoperatively in a large group of patients with colorectal neoplasia. A prospective study of 94 patients who underwent putative curative resection for colorectal carcinoma (CRC) was performed to ascertain if serum mutant KRAS2 could be used postoperatively as a disease marker.
Methods: Preoperative sera from 78 patients were analysed (group A). Sera from 94 patients were obtained three monthly for up to three years during the postoperative period (group B). Codon 12 and 13 KRAS2 mutations were analysed in matched tumour and serum samples.
Results: In the preoperative group (group A), KRAS2 mutation was found in 41/78 (53%) tumours and in 32/78 (41%) preoperative sera. Of 41 tumour KRAS2 mutation positive cases, 31/41 (76%) had an identical serum mutation detectable. In group B, the postoperative follow up group, 60/94 cases were primary tumour KRAS2 mutation positive. Of these 60, 16/60 (27%) became persistently serum mutant KRAS2 positive postoperatively. Ten of 16 (63%) of these developed a recurrence compared with only 1/44 (2%) patients who remained serum mutant negative (odds ratio 71.7 (95% confidence interval 7.7–663.9; p=0.0000). None of 34 tumour mutation negative cases became serum mutant KRAS2 positive postoperatively, despite recurrence in 9/34 patients. The relative hazard of disease recurrence in postoperative serum mutant KRAS2 positive patients was 6.37 (2.26–18.0; p=0.000).
Conclusions: Serum mutant KRAS2 can be detected preoperatively in all stages of colorectal neoplasia. Postoperatively, serum mutant KRAS2 is a strong predictor of disease recurrence, stronger even than Dukes' stage of disease, and thus shows potential for use in clinical practice as a marker of preclinical disease recurrence.
- DNA
- KRAS2
- KRAS2 mutation
- prognosis
- colorectal carcinoma
- CRC, colorectal carcinoma
- TVA, tubulovillous adenoma
- CEA, carcinoembryonic antigen
- OR, odds ratio
- PCR, polymerase chain reaction
Statistics from Altmetric.com
- CRC, colorectal carcinoma
- TVA, tubulovillous adenoma
- CEA, carcinoembryonic antigen
- OR, odds ratio
- PCR, polymerase chain reaction
Colorectal cancer (CRC) is the most common gastrointestinal cancer diagnosed in both the USA and Europe each year.1KRAS2 gene mutations arise early in the colorectal adenoma to carcinoma progression and over 50% of human CRCs and adenomata harbour KRAS2 mutations.2,3 Mutation within the KRAS2 gene occurs most commonly at positions 1 and 2 of codons 12 and 13.3–6
Molecular staging strategies using either immunohistochemistry or reverse transcription polymerase chain reaction (PCR) have shown that approximately 30–40% of patients who were histologically stage II/Dukes' B disease harbour occult micrometastases in the locoregional lymph nodes.7–10 Using similar strategies, micrometastases have also been detected in the bone marrow of patients with various gastrointestinal malignancies.11,12 Thus it is becoming evident that standard staging methods understage many patients with colorectal cancer and that the use of molecular techniques can detect micrometastatic disease in a significant proportion of patients with purported early disease.
Tumour derived mutant DNA has been detected in various body fluids. Mutations in various genes, including KRAS2, p53, and CD44 among others, have been detected in stool samples of patients with CRC.13–16 Mutant KRAS2 has been detected in DNA extracted from pancreatic fluid in patients with pancreatic carcinoma,17 from urine specimens in patients with bladder cancer,18 and from the sputa of patients with lung cancer.19
Tumour derived circulating mutant DNA is also detectable in the plasma or serum of cancer patients, as indeed is tumour derived RNA.20,21 Circulating wild-type DNA is detectable at low levels in normal healthy controls.22,23 Mutant circulating DNA has been found in the plasma or serum of patients with small cell lung cancer,24 head and neck cancer,25 clear cell renal cancer,26 pancreatic cancer,27 breast cancer,28 hepatocellular carcinoma,29 non-small cell lung cancer,30 and CRC.31–33 In almost all instances where tumour tissue was available, the mutation in the plasma was identical to that detected in the primary tumour, indicating that the circulating mutant DNA was of tumour origin. Anker et al reported that mutant KRAS2 was detectable in the plasma of 86% (6/7) of CRC patients in whom the KRAS2 mutation was present in the primary CRC.33 To date, studies investigating circulating mutant DNA in CRC patients have included relatively small numbers of patients, have examined only preoperative serum or plasma samples, and have not included many patients with early disease.
The present study was conducted to investigate if mutant KRAS2 could be detected in the preoperative sera of a large group of colorectal neoplasia patients ranging from early (dysplastic adenoma) to advanced (Dukes' D) CRC. Furthermore, a prospective postoperative follow up study of almost 100 patients with CRC who had undergone putative curative resection was performed to investigate if mutant circulating KRAS2 could be detected postoperatively and, if so, whether this was of prognostic significance, with potential use as a disease marker and predictor of disease recurrence.
MATERIALS AND METHODS
Patients
Ethics approval was obtained from our institution's ethics committee and all patients gave written informed consent prior to participation in the study. A total of 123 patients with a confirmed histological diagnosis of colorectal neoplasia (ranging from severely dysplastic tubulovillous adenoma (TVA) to Dukes' D colorectal cancer) were included in this study. All consecutive patients diagnosed with putative curative colorectal neoplasia (including non-endoscopically removable TVA patients) between January 1997 and March 1998 were approached to participate in the study. Seventy eight of 95 patients agreed to participate and were recruited to group A at the time of first diagnosis of the disease and preoperative blood samples were obtained in all cases. Group B comprised 94 patients including 49 from group A, who were followed from the time of surgery. A further 45 patients (of 60 randomly approached patients) who had been diagnosed with curable colorectal cancer at our institution in the preceding four years, all of whom still attended for routine surveillance, agreed to participate in the study. All patients in group B had undergone a putative curative resection for their disease. Twenty nine of 78 patients in group A did not enter the postoperative follow up part of the study for one of the following reasons: withdrew consent regarding further participation (n=9); died postoperatively (n=2); peroperative discovery that curative resection was impossible (n=5); or logistic reasons (n=13). Group B patients were followed for up to three years from the time of entry into until completion of the study (January 1997 to January 2000). Table 1 shows the clinical and demographic details of the patients in both groups.
Characteristics of patients and controls
The postoperative blood sampling protocol included a sample taken one week and one month postoperatively, followed by three monthly samples thereafter until the end of the study period. Patients in group B who entered the study some time after surgery had blood drawn every three months from the time of entry into until completion of the study. No patients were lost to follow up. During the study, all patients had standard modality disease follow up, including regular blood biochemistry, yearly colonoscopy, carcinoembryonic antigen (CEA) determination, yearly abdominal ultrasound or computed tomography scan, and yearly chest x ray. In the case of patients with TVAs, follow up was less stringent, consisting of colonoscopy at years 1 and 3 postoperatively.
Sera were obtained from 20 healthy controls, consisting of individuals who had undergone a normal colonoscopy for screening purposes or for investigation of lower gastrointestinal symptoms.
DNA extraction from serum and tumour samples
Blood (10 ml) was drawn into serum tubes (Becton-Dickinson SSAT tubes) and placed immediately on ice. Samples were kept on ice during all stages of handling. Within two hours, bloods were centrifuged at 1500 g for 30 minutes at 4°C. Serum was removed and stored at −20°C until further use. Serum 1 ml was used in patients and serum 2 ml in controls. Qiagen mini-kit blood DNA extraction kits were used to extract DNA from sera. The standard protocol was modified to include the addition of poly-A DNA carrier (Promega, Southampton, UK) facilitating better adsorption of DNA to the Qiagen column. DNA was eluted in a final volume of 65 μl. Control β-actin PCR with 5 μl of serum DNA was performed to confirm the presence of amplifiable quantities of DNA.
DNA was extracted from paraffin tissues using a standard technique.34 Control β-actin PCR was performed on all tumour derived DNA to ensure integrity of the extracted DNA.
KRAS2 codon 12 mutation analysis
Mutation at codon 12 was analysed using a combination of a previously described semi nested mutant enrichment technique33 and direct sequencing. The primers used were those described by Anker and colleagues.33 The upstream primer (p4) is modified to create a BstN1 (New England Biolabs, Beverly, USA) restriction site in the presence of wild-type but not mutant KRAS2 codon 12. An initial 15 cycles of PCR were performed. The 25 μl reactions contained 3 μl of serum DNA or 1 μl of tumour DNA, 15 mM Mg++, 0.9 pmol of each of primers p4 and p5, 0.08 μl Taq DNA polymerase (Promega), and 2 mM each of dNTP mix. The PCR product (3 μl) was digested for 12 hours with 20 units BstN1. The restriction digest product (3 μl) was then reamplified for 35 cycles in a 50 μl reaction. The reaction conditions for this second PCR reaction were 6 pmol of primer p4 and p6, 1.5 mM Mg++, and 0.1 μl Taq DNA polymerase, giving a final PCR product of 135 base pairs. A hot start was used in both PCR reactions, the cycle conditions were 94°C×4 minutes, 80°C×5 minutes, 15 or 35 cycles of (94°C×30 seconds, 55°C×60 seconds, 72°C×50 seconds), and then 72°C×4 minutes. In all reactions, two no template controls (sterile water), one wild-type DNA control (healthy control whole blood DNA), and one mutant positive control (DNA from SW 480 cell line) were included.
The final PCR product was then “cleaned up” by running the products on a 2% agarose gel; the 135 base pair band was excised, and DNA was purified and retrieved using a Qiaquick gel extraction kit, eluting in a final volume of 40 μl. Direct sequencing was performed on 15 μl of the purified PCR product using the 33P-USB Thermo Sequenase kit (USB, Cleveland, Ohio, USA). The sequencing primer (pQ) used was identical to that used by Anker and colleagues33 and sequenced the non-coding DNA strand. Sequencing products were electrophoresed on a 6% polyacrylamide gel at 70 W for two hours. Gels were dried, exposed to x ray film for 24 hours, and then developed.
KRAS2 codon 13 mutation analysis
A similar mutant enrichment technique to detect mutations at codon 13 of the KRAS2 gene was designed. An upstream primer was designed, p13 (5`- TATAAACTTGTGGTAGTTGGCCCTGGT-3`), which creates a Bgl1 (New England Biolabs) restriction site in the presence of wild-type but not mutant sequence. Eighteen cycles of initial PCR were performed. The 25 μl reaction conditions were: 1 μl of tumour DNA or 3 μl of serum DNA, 2.5 mM Mg++, 4 pmol each of primer p13 and p6, 0.08 μl Taq DNA polymerase (Promega), and 2.0 μl of 2 mM each dNTP. The PCR product (10 μl) was then digested for one hour at 37°C using Bgl1. The restriction digest mix (5 μl) was reamplified for 35 cycles using 6 pmol each of primer p13 and p6, 2.5 mM Mg++, 2.5 μl of 2 mM each dNTP, and 0.1 μl Taq polymerase. The hot start PCR conditions optimised for both the first and second PCR reactions were: 94°C×2 minutes, 80°C×5 minutes, 20 or 35 cycles of (94°C×30 seconds, 54°C×60 seconds, 72°C×60 seconds), and then extension at 72°C×4 minutes. The final 125 base pair PCR product was purified and sequenced using identical methods described for sequencing of the KRAS2 codon 12 product. In all experiments the following controls were included: two no template controls (sterile water), two mutant positive controls (DNA extracted from the hct-116 colorectal cancer cell line which harbours a heterozygous KRAS2 codon 13 aspartate mutation), and one wild-type DNA control.
Tumour DNA and serum DNA samples were first blindly analysed for mutation at codon 12, the most prevalent mutation. Tumour and serum samples were processed separately to avoid the possibility of cross contamination. Tumour DNA and serum DNA samples that were codon 12 mutation negative were then analysed for mutations at codon 13, again in a blinded manner. Serum samples from 20 healthy controls were analysed for KRAS2 codon 12 and 13 mutations.
Statistics
Results were analysed using the SPSS statistics package for Windows. χ2 analysis, descriptive statistics, and the McNemar χ2 test for related samples and Fisher's exact test were employed. Trend analysis was performed by χ2 analysis. Odds ratios (OR) with 95% confidence intervals (CI) were calculated as estimates of risk. Survival analysis was performed using Kaplan-Meier disease free survival analysis and log rank statistic. A Cox regression multivariate analysis was performed to evaluate the independent prognostic effects on survival in CRC of Dukes' stage, preoperative serum mutant KRAS2 positivity, postoperative serum mutant KRAS2 positivity, and adjuvant chemotherapy treatment.
RESULTS
Group A: preoperative group results (n=78)
Wild-type KRAS2 sequence was found in the tumour and serum samples of all colorectal neoplasia cases (n=78) and in all serum samples from controls (n=20). KRAS2 mutation at either codon 12 or 13 was identified in 41/78 (53%) of the tumour samples and in 32/78 (41%) of the serum samples (table 2). Of the 41 tumour KRAS2 mutation positive cases, 31/41 (76%) had an identical mutation present in serum (table 2). A further serum mutant KRAS2 positive result was found in 1/37 (3%) tumour KRAS2 mutation negative cases resulting in a total of 32/78 (41%) preoperative serum mutant positive cases (table 2).
Tumour and serum KRAS2 mutation and carcinoembryonic antigen (CEA) status in group A and controls
There was no significant relationship between CRC stage and the prevalence of mutant KRAS2 in serum. However, this was confounded by the fact that differing proportions of patients with each stage of disease were tumour KRAS2 mutation positive (table 2). However, in patients who were tumour KRAS2 mutation positive (n=41), there was a non-significant increase in the prevalence of serum mutant KRAS2 positivity with advancing disease stage (p=0.308, Fisher's exact test) (table 2). Some representative results of sequencing are shown in fig 1A.
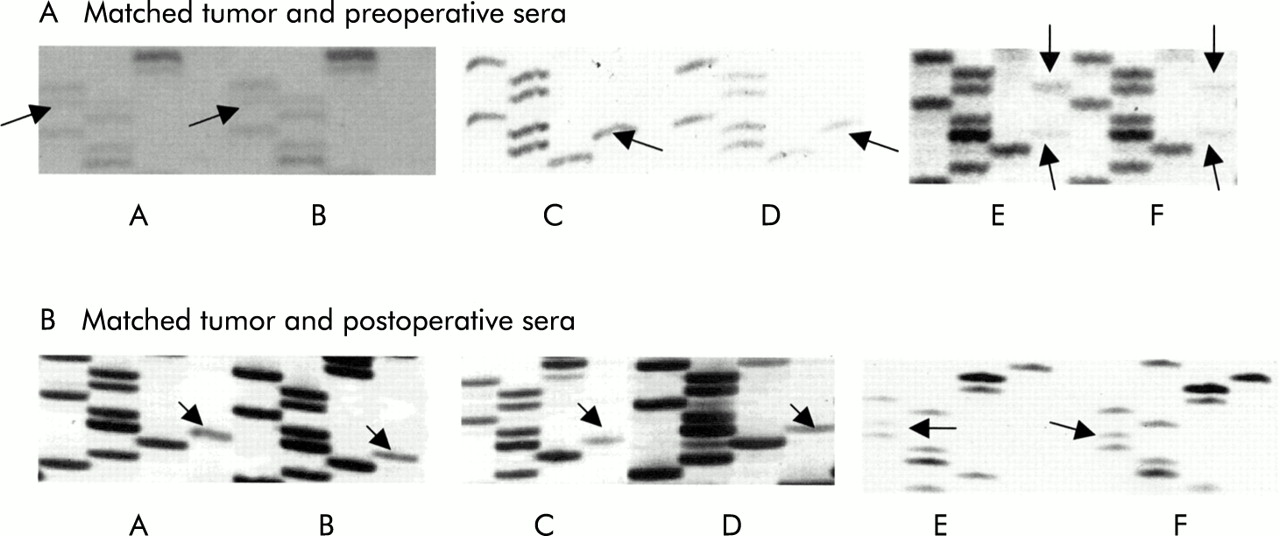
Representative results of sequencing of the region flanking codon 12 and 13 of the first exon of the k-ras gene. Mutant bands are indicated by arrows. Part (A): (A) Tumour DNA from Dukes' B case P1, with a codon 12 cysteine mutation. (B) Preoperative serum from case P1, showing an identical codon 12 cysteine mutation. (C) Tumour DNA from Dukes' C case P2, showing a codon 13 aspartate mutation. (D) Preoperative serum from case P2, with the mutation matching that of the primary tumour. (E) Tumour DNA from Dukes' A case P3 showing a double aspartate mutation in both codons 12 and 13. (F) Preoperative serum DNA from case P3 showing mutant codons 12 and 13, identical to that found in the tumour. Part (B): (A) Tumour DNA from Dukes' C case P4, with a codon 13 aspartate mutation. (B) Postoperative serum from case P4, with an identical codon 13 aspartate mutation. (C) Tumour DNA from Dukes' C case P5, with a codon 13 aspartate mutation. (D) Postoperative serum DNA from case P5, with a matched aspartate mutation to that in the tumour. (E) Tumour DNA from Dukes' B case P6, with a codon 12 valine mutation. (F) Postoperative serum DNA from case P6, with a codon 12 mutation, identical to that in the tumour.
The sensitivity of serum mutant KRAS2 detection was compared with CEA measurement in patients with colorectal neoplasia using McNemar's χ2 paired observations analysis (table 2). When all patients were included, there was no significant difference between the prevalence of serum KRAS2 positivity and raised CEA levels in any stage of disease. When the 41 tumour KRAS2 mutation positive cases were analysed separately, serum mutant KRAS2 analysis was statistically superior to CEA measurement as a marker of CRC. Six of 41 patients were negative for both tests, 5/41 were serum mutant KRAS2 negative/CEA positive, 16/41 were serum mutant KRAS2 positive/CEA negative, and 14/41 were positive for both tests (p=0.027, Fisher's exact test).
The prognostic value of a positive preoperative serum mutant KRAS2 test was evaluated, including only 49/78 patients from group A for whom postoperative follow up data were available as part of the group B study. Cox regression analysis showed that preoperative positive serum mutant KRAS2 was not an independent prognostic indicator for disease recurrence postoperatively (relative hazard 2.07, 95% CI 0.3–14.8; p=0.466).
Group B: results of postoperative follow up (n=94)
Sixty of 94 (64%) cases in group B were tumour KRAS2 mutation positive (tables 3, 4). A subgroup of patients from group B (85/94), comprising patients with Dukes' A, B, or C disease (excluding the six TVA and the three Dukes' D cases who had extremely low and high risks of recurrence, respectively) were analysed separately as these patients were of greater clinical interest and relevance in terms of recurrence detection. Fifty four of 85 (64%) patients in this group were tumour KRAS2 mutation positive (table 4). There was no significant difference in postoperative follow up duration between tumour KRAS2 mutation positive and mutation negative patients (table 1) for either the 94 or 85 patients (p=0.06 and p=0.061, respectively, Mann Whitney U test).
Tumour KRAS2 mutation status and disease stage of group B patients
Postoperative mutant serum KRAS2, tumour KRAS2 status, and recurrence in group B
When disease recurrence rates were compared in tumour KRAS2 mutation positive and tumour KRAS2 mutation negative cases, there was no significant difference in recurrence rates when either all 94 patients in group B (p=0.354) or only the 85 patients with Dukes' A, B, or C disease (p=0.654) were analysed (table 4).
Wild-type KRAS2 was found in all tumour and serum samples. A patient was considered postoperative serum mutant KRAS2 positive only if two consecutive serum samples were positive (three months apart). Figure 1B shows some representative KRAS2 sequencing results of matched tumour and postoperative serum samples. No patient was postoperative serum mutant KRAS2 positive on only one occasion. Serum taken one week after surgery was negative for mutant KRAS2 in 49/49 patients in whom this was tested.
When all 94 patients in group B were examined, in the tumour KRAS2 mutation negative group (n=34) none of the 9/34 recurrent cases became serum mutant KRAS2 positive during follow up (table 4, fig 2). By contrast, 16/60 (27%) tumour KRAS2 mutation positive patients became persistently serum mutant KRAS2 positive in the follow up period, of which 10/16 (63%) developed recurrent disease during the follow up period compared with only 1/44 (2%) who remained serum KRAS2 mutation negative (table 4, fig 2). Six of 16 (37%) patients who remained persistently serum KRAS2 mutation positive had not developed clinical disease recurrence by the end of follow up (table 6). Thus within tumour KRAS2 mutation positive patients (n=60), the OR for cancer recurrence was 71.6 (95% CI 7.7–663.9; p=0.0000) in serum mutant KRAS2 positive compared with serum mutant KRAS2 patients.

Graphical representation of the outcome of all patients in group B according to tumour and serum KRAS2 mutation status. These results are presented in tabular form in table 4.
Separate analysis of the subgroup of 85 patients from group B with Dukes' A, B, or C disease gave similar results to those of all 94 patients (table 4). In this subgroup the OR for cancer recurrence in tumour KRAS2 mutant positive/serum KRAS2 mutant positive patients compared with tumour KRAS2 mutant positive/serum KRAS2 mutant negative patients was 57 (95% CI 6.1–534.5; p=0.0000).
A Cox regression analysis was performed to investigate the independent prognostic value of postoperative serum mutant KRAS2 as a predictor of disease recurrence (table 5). For this model only the 85/94 patients with Dukes' A, B, or C disease were included. The independent effects of postoperative serum mutant KRAS2 positivity, Dukes' stage, and adjuvant chemotherapy were investigated (table 5). Because of the low number of events recorded in patients with Dukes' A CRC, for the Cox regression analysis Dukes' A and B patients were analysed together and compared with patients with Dukes' C CRC. Positive serum mutant KRAS2 was associated with a relative hazard of 6.37 (95% CI 2.3–18.0; p=0.000), increasing Dukes' stage had a relative hazard of 3.1 (95% CI 0.96–9.9; p=0.056), and adjuvant chemotherapy treatment was associated with a relative hazard of 0.47 (95% CI 0.12–1.8; p=0.278) for disease recurrence. Figure 3 shows Kaplan Meier disease free survival curves for both tumour mutant KRAS2 positive and negative patients (n=85) and for tumour KRAS2 mutation positive patients separately (n=54).
Cox regression analysis of factors influencing risk of disease recurrence

{kind=link}
{kind=link}
{kind=link}
Kaplan Meier disease free survival curves according to serum mutant KRAS2, tumour mutant KRAS2, and disease stage. Survival analysis included only patients with Dukes' A, B, or C disease, as inclusion of patients with tubulovillous adenoma or Dukes' D may bias the results because of the low and high recurrence rates, respectively. Below, the symbols (+) and (−) refer to KRAS2 mutation status. (A) In n=85 patients, tumour (+) and (−), survival was significantly worse in serum (+) patients (p=0.0000). (B) In tumour (+) patients (n=54), survival was significantly worse in serum (+) patients (p=0.0000). (C) Stratification of n=54 patients who were tumour (+) on the basis of disease stage showed that serum (+) patients had a significantly worse prognosis in each disease stage (p=0.0000 for early disease and p=0.007 for late disease). The disease free survival curves were similar for patients who were serum (+) and who had either early or late disease and for patients who were serum (−) with either early or late disease.
Including all 94 patients irrespective of tumour KRAS2 status, detection of postoperative serum mutant KRAS2 was 52.6% sensitive and 92% specific for disease recurrence, with a positive predictive value of 62.5%. For the 60 tumour KRAS2 mutation positive patients, detection of postoperative serum mutant KRAS2 was 91% sensitive and 88% specific for disease recurrence, and the positive predictive value for disease recurrence was 62.5%.
Serum mutant KRAS2 positivity was compared with standard modality disease surveillance as a marker of disease recurrence (table 5). Seven of 10 (70%) patients who were serum mutant KRAS2 positive and who developed recurrence were serum mutant KRAS2 positive before the detection of disease recurrence using standard modalities. Three of 10 patients had concomitant elevated CEA at the time of first detection of positive serum mutant KRAS2 (table 6). The median “lead time” was four months, ranging from 0 to 16 months prior to diagnosis using standard surveillance methods.
Site and timing of recurrence in postoperative serum mutant K-ras positive patients
DISCUSSION
We have shown that mutant KRAS2 was detectable preoperatively in the serum of 32/78 (41%) patients with colorectal neoplasia, confirming the results of several smaller studies.31–33 This is the largest series of colorectal neoplasia patients to be investigated for preoperative circulating mutant DNA. In all but one case the serum KRAS2 mutation was identical to that detected in the primary tumour, indicating that the serum mutant DNA was of tumour origin. Sampling error of the paraffin embedded tumour specimen in this discrepant case may account for failure to detect a mutation within the tumour.
Preoperatively, detection of mutant circulating KRAS2 was 100% specific for colorectal neoplasia in this study. The positive predictive value for colorectal neoplasia was 100% but sensitivity was relatively low at 41%. The main factor limiting the sensitivity of serum mutant KRAS2 detection was the prevalence of KRAS2 mutation within the primary tumour: only 41/78 (53%) of all colorectal neoplasias had a KRAS2 mutation in the primary tumour but 31/41 (76%) of these tumour KRAS2 mutation positive cases had an identical mutation detectable in the circulation. These results indicate that circulating serum mutant KRAS2 is a marker of the presence of tumour but only when the tumour harbours a KRAS2 mutation.
Circulating mutant KRAS2 was detectable even in patients with TVA (large TVAs requiring surgical excision) in this study: 4/9 (44%) of all TVAs, or 4/7 (57%) KRAS2 mutation positive TVAs, had detectable circulating mutant KRAS2. This concurs with the findings of a recent study that demonstrated mutant serum KRAS2 in patients with colonic adenomata, but in contrast with our current study the previous study did not have matched tumour and serum samples in all cases.35
Detection of mutant circulating DNA in patients with colonic adenomata is significant for two reasons. Firstly, it provides insight into the mechanism of circulating DNA in patients with malignancy. It has been proposed that mutant DNA might enter the circulation of cancer patients through direct release from circulating metastatic tumour cells or from the in situ lesion.36 As premalignant lesions have no metastatic potential, the serum mutant KRAS2 detected in these cases must be released from the in situ lesion. Secondly, the ability to detect premalignant colorectal neoplasms through a serum assay has implications for screening. At present, a major limiting factor for the use of this assay for screening purposes is the low sensitivity but this could be greatly enhanced by analysis of multiple genes commonly mutated in CRC, such as APC and p53. Indeed, detection of multiple colon cancer specific DNA changes in stool samples has recently been shown to increase the sensitivity of cancer detection above that found when using only one marker.16 Thus a combination approach could provide a serum detectable somatic genetic marker for up to 100% of colorectal neoplasias.
This is the first study to prospectively investigate the natural history of circulating mutant DNA following resection of the primary tumour in patients with CRC. Strikingly and of great potential clinical relevance, our results indicate that the presence of postoperative serum mutant KRAS2 is strongly predictive of disease recurrence in CRC patients. Indeed, in a multivariate analysis, positive postoperative serum mutant KRAS2 was the strongest predictor of CRC recurrence, stronger even than the influence of Dukes' stage or of treatment with adjuvant chemotherapy. This is consistent with the findings of several small studies of patients with small cell lung cancer,37 non-Hodgkin lymphoma,38 and pancreatic cancer,39 where persistence or reappearance of mutant DNA in the serum post-treatment heralded a poor prognosis. As with the preoperative results, the main factor limiting the performance of serum mutant KRAS2 as a marker of CRC recurrence was the presence or absence of a KRAS2 mutation in the primary tumour. This is keeping with the previously reported finding that KRAS2 status of a malignant clone is stable throughout its natural history.40
All positive patients who entered the study at a later stage (not immediately post-surgery), tested positive when first tested, and thus are likely to have been positive for some time prior to this. However, accurate documentation of the time at which they became positive is not possible. None of the one week post-surgery serum samples (n=49) had detectable mutant KRAS2. This is despite the fact that some of these patients subsequently became serum mutant KRAS2 positive during the follow up period, indicating that they must have had occult tumour present even at one week post-surgery. This suggests two things: firstly, that the half life of circulating tumour DNA in serum is less than one week, which is not surprising as plasma contains large amounts of DNAses that are likely to continuously degrade DNA present in plasma such that the presence of DNA in plasma represents an equilibrium between release from body cells and degradation by DNAses. Secondly, this finding also suggests that a critical mass of tumour must be present before sufficient mutant DNA is released into the circulation allowing it to be detectable by the methods employed in the current study. Improved sensitivity enabling detection of minute quantities of circulating mutant KRAS2 may be facilitated by the use of more modern technology such as DNA microchips.
A single patient (M39 in table 6) who was postoperatively serum mutant KRAS2 positive subsequently developed a local recurrence and underwent a second curative resection; the subsequent serum mutant KRAS2 assay was negative. This demonstrates that serum mutant DNA analysis can detect clinically treatable recurrent disease and not merely identify untreatable metastatic disease at an earlier stage. Furthermore, we identified six patients who were serum mutant KRAS2 positive post-surgery in whom clinical disease recurrence has not yet developed. It is likely that these patients have occult disease recurrence and thus close follow up is imperative. Several other explanations for this finding are also possible. They may have a colonic polyp which was missed at colonoscopy and which harbours the same KRAS2 mutation as the primary resected tumour. It has been shown that mutant serum KRAS2 is detectable even in cases of small colonic polyps.35 Secondly, molecular field effect changes are found in histologically normal colonic mucosa in patients with colonic tumours41 and this could potentially release mutant DNA into the circulation. Finally, a synchronous tumour at another site might be present in one or more of these patients. However, the fact that in all cases the postoperative serum KRAS2 mutation was identical to that in the primary tumour suggests that the serum mutant DNA originates from the same clone of cells as the primary tumour. Further study and follow up of these and other patients will help elucidate this issue.
In this study, the disease recurrence rate in tumour KRAS2 mutation negative patients (26%) was not significantly different (p=0.354) to that in tumour KRAS2 mutation positive patients (18%). However, the follow up duration of the tumour KRAS2 mutation group was slightly shorter than that of the tumour KRAS2 mutation positive group. This difference in follow up duration may disguise an otherwise significant difference in recurrence rate between these two groups.
The findings of our study are novel. We have shown that serum mutant DNA can be detected preoperatively in a large proportion of patients with colorectal neoplasia, including those with premalignant disease. Furthermore, while circulating mutant KRAS2 appears to be a useful marker of cancer only in patients who have a KRAS2 mutation in the primary tumour, sensitivity could be greatly enhanced by analysing for multiple mutations and by using DNA chip technology.42 Significantly, we showed for the first time that postoperative serum mutant KRAS2 detection is a highly significant predictor of disease recurrence, a stronger predictor even than Dukes' stage. As a disease marker, postoperative serum mutant DNA detection has great potential in the follow up management of colorectal and other cancers. In particular, postoperative mutant DNA detection might prove useful in identifying patients with early occult disease recurrences who might benefit from early intervention with chemotherapy.
Acknowledgments
This work was supported by a grant from PPP Healthcare Medical Trust, a charitable organisation, to Barbara Ryan. Ross McManus is a Wellcome Fellow and Jacqueline Daly was supported by a grant from the Irish Health Research Board. This work was presented in abstract form at the Digestive Disease Weeks in San Diego 2000 and Atlanta 2001.