Article Text

Abstract
Background: Autoantibodies against transglutaminase 2 (TG2) are thought to be responsible for the endomysial (EMA), reticulin (ARA), and jejunal antibody (JEA) tissue binding of serum samples from coeliac patients but the exclusive role of TG2 in these staining patterns has not yet been established.
Aims: To evaluate whether antigens other than TG2 contribute to EMA/ARA/JEA reactions.
Patients: Serum samples from 61 EMA/ARA/JEA positive untreated patients with coeliac disease, 40 dermatitis herpetiformis patients, and 34 EMA/ARA/JEA negative non-coeliac controls were tested.
Methods: TG2 knockout (TG2−/−) and wild-type mouse oesophagus, jejunum, liver, and kidney sections, and TG2−/− sections coated with human recombinant TG2 were used as substrates in single and double immunofluorescent studies for patient IgA binding and tissue localisation of TG2, fibronectin, actin, and calreticulin.
Results: None of the patient serum samples elicited EMA, ARA, or JEA binding in TG2−/− morphologically normal tissues. In contrast, 96 of 101 gluten sensitive patient samples (95%) reacted with wild-type mouse tissues and all 101 reacted in EMA/ARA/JEA patterns with TG2−/− mouse tissues coated with human TG2. Serum IgA binding to TG2−/− smooth muscle cells was observed in low titres in 31.1%, 27.5%, and 20.5%, and to TG2−/− epithelium in 26.3%, 5.0%, and 8.8% of coeliac, dermatitis herpetiformis, and control samples, respectively. These positivities partly colocalised with actin and calreticulin but not with TG2 or fibronectin.
Conclusions: EMA/ARA/JEA antibody binding patterns are exclusively TG2 dependent both in coeliac and dermatitis herpetiformis patients. Actin antibodies are responsible for some positivities which are not part of the EMA/ARA/JEA reactions.
- coeliac disease
- dermatitis herpetiformis
- endomysial antibodies
- transglutaminase 2
- knockout mouse
- ARA, R1-type reticulin antibodies
- ELISA, enzyme linked immunosorbent assay
- EMA, endomysial antibodies
- JEA, jejunal antibodies
- TBS, Tris buffered saline
- TG2, transglutaminase type 2 or tissue transglutaminase
Statistics from Altmetric.com
- ARA, R1-type reticulin antibodies
- ELISA, enzyme linked immunosorbent assay
- EMA, endomysial antibodies
- JEA, jejunal antibodies
- TBS, Tris buffered saline
- TG2, transglutaminase type 2 or tissue transglutaminase
In a subgroup of individuals with HLA-DQ2 or DQ8 alleles, cereal gluten ingestion induces coeliac disease, which is accompanied by circulating autoantibodies against certain extracellular matrix structures.1 These autoantibodies are highly pathognomic for untreated coeliac disease, are predominantly of the IgA class, and disappear on treatment with a gluten free diet. They also occur in cases of dermatitis herpetiformis where gluten sensitivity is manifested as both intestinal and skin disease.2 According to the tissue antigen used and the typical binding pattern observed, the coeliac autoantibodies have been named R1 type reticulin (ARA), endomysial (EMA), and jejunal (JEA) antibodies.1
Only since 1997 was it discovered that EMA react with the enzyme transglutaminase 2 (TG2, tissue transglutaminase), one of the eight currently known transglutaminase enzymes which form epsilon-gamma protein cross links between glutamine and lysine residues of proteins.3,4 Subsequently, TG2 based enzyme linked immunosorbent tests (ELISA) of high diagnostic value for coeliac disease were developed.5 Monoclonal antibodies against TG2 also bind to tissue sections in EMA, ARA, and JEA patterns under conditions similar to those in indirect immunofluorescent tests used in clinical diagnostics.5–7 Moreover, soluble TG2 from the guinea pig added to tissue fibronectin can replace the chemically removed endomysial target antigen which appears to be bound to fibronectin.7 However, a question remains as to whether other autoantigens contribute to the endomysial and related tissue binding patterns of coeliac serum samples. Autoantibodies against actin, calreticulin, desmin, and a 90 kDa dermal glycoprotein have also been described in coeliac patients.8–11 Furthermore, Uhlig et al reported that not all coeliac antibody binding sites match with TG2.12
In the present study we aimed to establish whether coeliac disease and dermatitis herpetiformis patient sera, giving the typical coeliac-type tissue staining pattern in normal primate and rodent tissues, contain autoantibodies to structures other than TG2. We took advantage of the recent development of TG2 knockout (TG2−/−) mice, as these mice are viable and have no major organ abnormalities.13 Using such TG2 deficient tissue sections, all antibody binding other than that elicited by TG2 should remain visible.
METHODS
Patient samples
IgA class EMA positive serum samples from 61 patients with untreated coeliac disease (aged 0.9–57.8 years; median 5.6) and from 40 patients with untreated dermatitis herpetiformis (aged 2.5–26.1 years; median 9.3), diagnosed according to current criteria by jejunal biopsy and skin immunofluorescent study, were investigated. Thirty four EMA negative serum samples from non-coeliac individuals with normal jejunal morphology (aged 1.2–60 years; median 6.8) were used as controls. Seven of these control serum samples were positive for IgA antibodies against gliadin. Serum samples were used with the permission of the ethics committee of the Heim Pál Children’s Hospital, Budapest.
Immunofluorescent studies
Oesophagus, jejunum, liver, and kidney tissues from TG2−/− C57BL/6 knockout mice13 and corresponding wild-type mice (TG2+/+) were snap frozen and kept at −70° C until testing. Unfixed frozen sections were used as substrates in indirect immunofluorescent studies with single and double labelling of patient IgA antibodies (green) and monoclonal mouse IgG1 class anti-TG2 antibodies CUB7402 and TG100 (red) (NeoMarkers, Fremont, California, USA), as previously described.7 Bound patient IgA antibodies were detected with fluorescein isothiocyanate conjugated secondary antibodies (Dako A/S, Glostrup, Denmark; diluted 1:120). Mouse tissue bound mouse anti-TG2 antibodies were detected with rabbit antimouse IgG1 (research antibody from Pharmacia Diagnostics AB, Uppsala, Sweden) and rhodamine conjugated swine antibodies against rabbit immunoglobulins (Dako), both diluted 1:100, to reduce background staining.
Serum samples from patients were initially tested in dilutions of 1:5 and 1:50 and, if necessary, titrated in serial twofold dilutions until the end point was reached. In double immunostainings, five coeliac, three dermatitis herpetiformis, and five control samples were used together with the commercial monoclonal anti-TG2 antibodies.
The EMA binding pattern was evaluated on the muscular layers of the oesophagus and jejunum, and on blood vessels, the ARA binding pattern on liver and kidney, and the JEA binding pattern on jejunal villi, as described elsewhere.7
In control double staining experiments, polyclonal rabbit antibodies against fibronectin (Dako; diluted 1:120), calreticulin (Alexis Biochemicals, San Diego, California, USA; diluted 1:200), and monoclonal mouse IgG2a antibodies against smooth muscle actin (MS-113; NeoMarkers; diluted 1:400, plus rabbit antibodies against mouse IgG2a; Zymed, San Francisco, California, USA) recognised with rhodamine conjugated antirabbit antibodies comprised the red component and human IgA binding (recognised as before) the green. In other control experiments, TG2−/− tissues were incubated prior to the indirect immunofluorescent study with soluble human recombinant TG2 (Pharmacia), 2 μg/ml in 0.05 M Tris, 0.15 M NaCl, pH 7.4 (Tris buffered saline (TBS)) with 0.005 M CaCl2 for 30 minutes.
Serum antibodies were also investigated on primate tissues, monkey oesophagus, normal human jejunum, and human appendix, respectively, as previously described.14,15
ELISA tests
IgA serum antibodies against gliadin and natural human TG2 antigen purified from normal red blood cells were measured by ELISA, as described elsewhere.16,17 The cut off level for TG2 antibody positivity was previously established at 10% of the absorbance of an international reference sample.5,18 IgA antibodies against calreticulin were investigated as follows: 0.2 μg/well purified calreticulin (Sigma Chemicals, St Louis, Missouri, USA) were coated overnight at 4°C on ELISA plates (Nunc-Immuno Maxisorp, Nunc, Roskilde, Denmark) in 0.03 M NaHCO3 buffer, pH 9.6. After three washes with TBS containing 0.1% Tween 20, patient samples diluted 1:100 in TBS were incubated for one hour. After extensive washing, bound IgA antibodies were measured with peroxidase conjugated rabbit antibodies against human IgA (Dako), similar to TG2 antibody detection. Commercial antibodies against calreticulin (Alexis) were used as positive controls.
Statistical differences between study groups were estimated using the Student’s t test and χ2 test, with two tailed probabilities. The median was used to characterise discontinuous antibody titres in immunofluorescence.
RESULTS
Coeliac-type positivity
On wild-type mouse tissues, serum samples of both coeliac and dermatitis herpetiformis patients but not controls showed EMA-, ARA-, and JEA-type IgA binding (table 1). The positivity clearly colocalised with the tissue distribution of TG2 shown by monoclonal TG2 specific antibodies (fig 1A, 2A). In contrast, in TG2 knockout mouse tissues, no EMA-, ARA-, or JEA-type binding was seen in any of the coeliac, dermatitis herpetiformis, or controls samples, even at low serum dilutions of 1:5. Similar results were obtained with IgG antibodies from IgA deficient coeliac patients (data not shown). No tissue positivity was seen with the CUB7402 (fig 1B, 2B) or TG100 monoclonal TG2 specific antibodies in TG2−/− knockout mice, thus confirming that they lack TG2. Tissues of TG2−/− mice were of normal morphology, and also the small intestinal mucosa, showing a villus height-crypt depth ratio of more than 5.
Coeliac-type endomysial (EMA), reticulin (ARA), and jejunal antibody (JEA) binding of serum samples from gluten sensitive and control patients to transglutaminase 2 knockout (TG2−/−) and corresponding wild-type (TG2+/+) mouse tissues

Both coeliac patient IgA (green inserts) and transglutaminase 2 specific monoclonal antibody CUB7402 (TG2, red inserts) bound to wild-type mouse oesophagus in identical endomysial patterns merging to yellow at colocalisation (A) but there was no specific binding to TG2 knockout mouse oesophagus (B). If human TG2 was inserted into TG2 knockout tissues, binding of both antibodies was similar to that in wild-type tissues (C). (Bar 20 μm.)
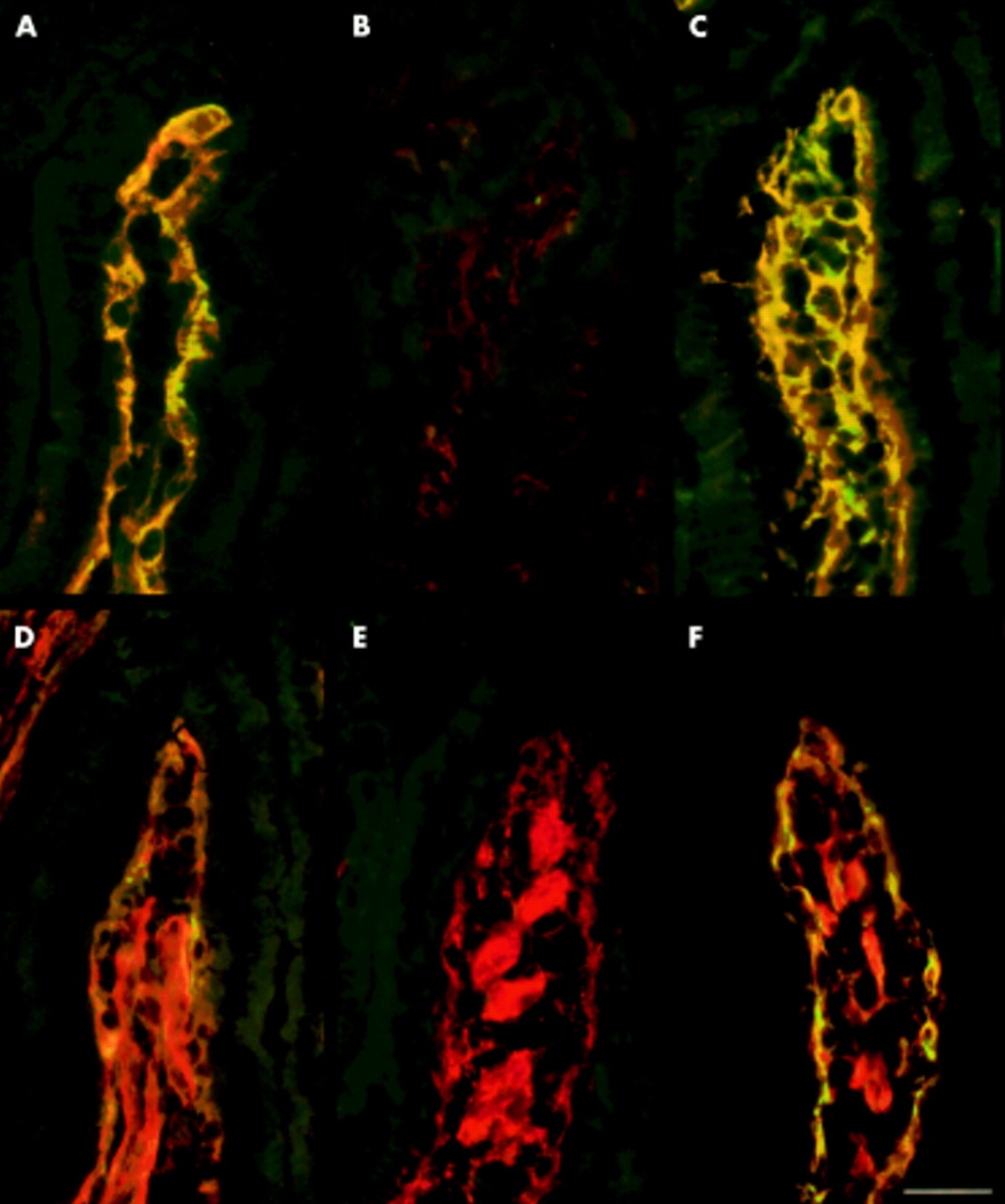
Wild-type (A, D) and transglutaminase 2 knockout mouse (B, C, E, F) jejunum double stained with coeliac patient IgA (green) and monoclonal transglutaminase 2 specific (TG2) antibodies (red) (A, B, C), and with coeliac patient IgA (green) and antibodies to fibronectin (D, E, F). Coeliac IgA binding completely colocalised with tissue TG2 (A) and partially colocalised with fibronectin (D) in wild-type jejunum and in TG2 knockout mouse tissues incubated with human recombinant TG2 (C, F). Coeliac IgA and TG2 monoclonal antibodies did not bind to TG2 knockout jejunum (B) where fibronectin was normally present (E). (Bar=30 μm.)
Fibronectin, the main protein anchoring TG2 to the extracellular matrix, was found in both TG2+/+ and TG2−/− tissues (fig 2D, E). However, in TG2 deficient mice it did not bind IgA from sera of coeliac or dermatitis herpetiformis patients (fig 2E).
To further confirm that EMA positivity elicited by serum samples from coeliac disease and dermatitis herpetiformis patients is TG2 dependent, we incubated TG2−/− tissues with recombinant human TG2 to insert TG2 into empty fibronectin binding sites.7 When these TG2 coated sections were used as substrates, both coeliac and dermatitis herpetiformis patient samples, as well as monoclonal TG2 specific antibodies, showed typical EMA-, ARA-, and JEA-type binding (fig 1C, fig 2C, F).
A total of 57 (93.4%) of 61 coeliac samples and 39 (97.5%) of 40 dermatitis herpetiformis samples showed clear coeliac-type positivity with all four rodent TG2+/+ organs. Two additional coeliac serum samples were positive only for the oesophagus. However, all 101 samples reacted in the EMA, ARA, and JEA pattern with TG2−/− mouse tissues coated with human TG2 and also reacted with natural TG2 from human red blood cells in ELISA (table 1). These findings demonstrate that some coeliac patients have only human TG2 specific antibodies.
Other immunofluorescent binding patterns in TG2 deficient mouse tissues
To investigate how frequently antibodies against structures other than endomysium/reticulin occur in coeliac or dermatitis herpetiformis patients, we evaluated all (even low titre) positivities seen in TG2−/− tissues and compared the results with non-coeliac controls.
Binding of patient IgA to smooth muscle cells or jejunal epithelial cells was frequently observed in TG2−/− tissues with both coeliac and dermatitis herpetiformis samples at a low (1:5) serum dilution but disappeared rapidly with further dilution (table 2). Median titres for both of these binding types were 1:5 while the median titre of the specific coeliac positivity on TG2+/+ tissues was 1:400 of the same samples. The reaction with smooth muscle cells was similarly prevalent in non-coeliac control serum samples (table 2), with considerable overlap in 95% confidence intervals (CI): coeliac samples 19.4–42.9%; dermatitis herpetiformis samples 13.5–41.5%; and controls: 6.7–34.4%.
Non-endomysial-type autoantibody binding patterns observed in transglutaminase 2 knockout mouse tissues with serum samples from gluten sensitive and control patients
With five coeliac serum samples, a positivity along the axis of the TG2−/− villi occurred, corresponding to the contractile bundles connected to the tunica muscularis mucosae layer (fig 3A, B). This binding pattern was clearly different from the coeliac-type EMA and JEA binding and showed colocalisation with smooth muscle actin in double immunostainings (fig 3C). These actin antibodies seemed to merge with EMA positivity when the same samples were tested on wild-type mouse jejunum (fig 3D, arrow). However, longitudinal or cross sections of the smooth muscle bundles which colocalised with actin (fig 3E) remained discordant in double immunostainings with TG2 (fig 3F). Actin bundles were similarly arranged in the human jejunum (fig 3G) but double stainings for actin and for coeliac binding with coeliac serum samples containing only EMA gave clearly distinct positivities (fig 3G). These findings indicate that actin reactivity is not part of the classical coeliac-type positivity.

{kind=link}
{kind=link}
{kind=link}
(A, B) Serum sample from a coeliac patient containing IgA antibodies (green) which bound to transglutaminase 2 knockout (TG2−/−) mouse jejunum along the axis of the villi (arrows) and colocalised to yellow with smooth muscle actin stained in red (C). Note the absence of endomysial (EMA) or jejunal (JEA) antibody staining. In wild-type mouse jejunum (D), the axial positivity merged with the IgA coeliac-type JEA of the same serum sample (arrow), colocalised with actin stained in red (E) but its cross sections remained discordantly green (arrows) after double staining with TG2 in red (F). (G) Actin bundles (red) were similarly arranged in the human jejunum but did not merge with the typical coeliac-type jejunal positivity (green) elicited by a coeliac patient serum sample containing only EMA. (Bar=30 μm.)
IgA antibodies which bound to epithelial cells in the TG2−/− jejunum occurred in 26.3% of coeliac samples (95% CI 15.1–37.4), a significantly higher proportion than in dermatitis herpetiformis (5.0%, 95% CI 0–11.8; p=0.002) or control patients (8.8%, 95% CI 0–18.5; p=0.02) (table 2). This positivity seemed to colocalise with immunoreactivity for calreticulin. However, mean reactivity of the samples with and without such epithelial staining was no different when serum antibodies to calreticulin were measured in ELISA (optical densities 0.235 (SD 0.162) v 0.197 (SD 0.176)). Of serum samples from controls, only those with gliadin antibody positivity reacted with epithelial cells, but mostly also with smooth muscle cells. There was however no statistically significant correlation between epithelial positivity, calreticulin, and gliadin antibody levels (data not shown).
In TG2−/− tissues, we also searched for additional binding patterns indicating possible cross reaction of coeliac (EMA) antibodies with other structures or with other transglutaminases in the absence of TG2. However, specific differences between controls and patient (sub)groups, including dermatitis herpetiformis patients, were not found.
DISCUSSION
Coeliac disease is an autoimmune disorder characterised by production of a large array of antibodies against tissues structures, matrix, and cellular antigens.1,3,8–11,19 According to the present findings, TG2 specific autoantibodies would appear to be of primary importance. Our results with TG2 knockout tissues for the first time directly prove that the diagnostically relevant EMA, ARA, and JEA binding patterns in serum samples from both coeliac and dermatitis herpetiformis patients are clearly and exclusively TG2 autoantibody dependent and target the fibronectin bound extracellular TG2 in normal human and rodent tissues. In the large series of serum samples we tested on TG2 knockout mouse tissues, TG2 unrelated non-specific EMA positivities were not observed. Complementing these results, we also experienced the same with EMA positive subjects in whom coeliac disease remained unconfirmed (data not shown). Thus the typical coeliac-type immunofluorescent positivity has an outstanding specificity for TG2 antibodies. Non-TG2 antibodies that produce EMA binding, albeit theoretically possible, seem to be very rare.
Serum samples from some seriously ill coeliac patients often show complex antibody binding. Due to high background binding to epithelial or smooth muscle cells, such “difficult” samples may appear negative for EMA in low serum dilutions20 but react with TG2 in ELISA. Smooth muscle cells contain high amounts of TG2. However, our results do not support the notion that reactivity with smooth muscle cells represents antibodies to intracellular TG2, as it was similarly observed in TG2 deficient tissues. We identified a subtype of smooth muscle antibodies as antibodies to actin. When present together with EMA, actin antibodies bound to the contractile bundles of the villi may modify the appearance of the coeliac-type positivity on the jejunum and may be responsible for the additional non-TG2 related components observed by other investigators.12 However, actin antibodies alone were not able to elicit EMA-type positivity in the absence of TG2.
We frequently found non-TG2 related antibodies in serum samples from coeliac and dermatitis herpetiformis patients but in low titres compared with endomysial autoantibodies. Apart from antibodies against actin, their binding lacked any specific morphological appearance and they occurred in less than half of all patients. Gliadin antibodies were associated both in coeliacs and controls with a higher rate of antibody binding to jejunal epithelial cells. These features support the view that these antibodies may be secondary and variably induced by tissue damage facilitating increased presentation of different self antigens to the immune system.19
As we now observed, coeliac disease specific autoantibodies react primarily with human TG2. Cross reaction with mouse TG2 was prevalent (93.4%) but not universal in our coeliac patients. The slightly different rate of coeliac autoantibody positivities in primate and rodent tissues was for a long time a subject of diagnostic debate and led to the misconception that ARA (rodent specific) and EMA (primate specific) tissue autoantibodies represent different antibody populations.21 However, with demonstration that a recombinant human TG2 protein inserted into mouse tissues comprises as good a coeliac autoantigen as natural primate tissues, we provide additional evidence that the “reticulin” and “endomysial” binding patterns are tissue-type and not species dependent detection methods of the same TG2 autoantibodies.
Our current understanding of the differences in clinical outcome between classical coeliac disease and dermatitis herpetiformis, where apart from gluten enteropathy dermal IgA deposits also occur, remains incomplete. It has been hypothesised that dermal IgA may represent immune complexes or additional antibodies against dermal proteins.2 Recent interest has focused on potential cross reaction with other structurally related transglutaminases, of which several are expressed in the epidermis (TG1, TG3, TG5, TG7).4,22,23 While serum antibodies from coeliac and dermatitis herpetiformis patients do not recognise TG1, it has been shown recently that a subset in both diseases cross reacts with TG3 (epidermal transglutaminase).23 In dermatitis herpetiformis, patients also develop high avidity antibodies specific for TG3, and TG3 is a component of dermal deposits and seems to be the major autoantigen involved in skin manifestations.23 However, dermatitis herpetiformis patients also produce autoantibodies that primarily react with TG2 and relate to the enteropathy. As we have shown here, TG3 specific antibodies do not contribute to the EMA positivity of these sera which requires targeting of TG2.
In similar ways, other particular presentation types of coeliac disease may potentially be associated with targeting of both TG2 and other transglutaminases. Studies with mRNA suggest that TG5,7 as well as TG3 are widely distributed in various tissues4,22,23 but the protein amounts and their histochemical localisation in non-dermal tissues are at present still unknown. Tissues of TG2 knockout mice are particularly suitable in the search for such autoantibody targets because the apparently normal condition of these mice is most probably maintained by other upregulated transglutaminases. 13 As we also observed, these mice do not develop jejunal villous atrophy even in the absence of TG2 function. However, with the conventional immunofluorescent technique, we did not find any staining differences between samples from coeliac and dermatitis herpetiformis patients. This is not in contrast with previous results because the autoantibodies in both conditions can cross react with TG3 in ELISA and they differ only in their relative avidity and specificity23 which, however, may not lead to visible differences in immunofluorescence. We were also unable to demonstrate staining differences between patients and controls in TG2 deficient tissues. Several explanations are possible, such as the small fraction of TG3 reactive autoantibodies23 or the low amounts of TG3 protein. In fact, TG3 specific rabbit antibodies, working in western blot or on skin sections, also failed to show jejunal immunofluorescent positivity in other studies.22,23 Therefore, further search for additional transglutaminase targets will necessitate more sensitive detection tools or unmasking of antigens.
In conclusion, our results confirm the identity of EMA, ARA, and JEA with TG2 specific autoantibodies and directly prove the exclusive role of TG2 in the specific and diagnostic coeliac-type immunofluorescent reactions.
Acknowledgments
The authors thank Gerry Melino (Biochemistry Laboratory, IDI-IRCCS, Department of Experimental Medicine and Biochemical Sciences, University of Rome, “Tor Vergata”, Rome, Italy) for the generous donation of TG2 knockout and wild-type mice. The Coeliac Disease Study Group is supported by the Päivikki and Sakari Sohlberg Foundation, the Foundation of the Friends of the University Children’s Hospitals in Finland, the Medical Research Fund of Tampere University Hospital, and the Academy of Finland Research Council for Health, funding decision number 73489. Ilma Korponay-Szabó was a recipient of travel grants from the European Science Foundation Transglutaminases and Protein-Cross Linking Programme and the Finnish Coeliac Society. This study was also funded by the Commission of the European Communities, specific RTD programme “Quality of Life and Management of Living Resources”, QLRT-1999-00037, “Evaluation of the prevalence of coeliac disease and its genetic components in the European population”. It does not necessarily reflect its views and in no way anticipates the Commission’s future policy in this area.