Article Text

Abstract
Background: Overproduction of colonic oxidants contributes to mucosal injury in inflammatory bowel disease (IBD) but the mechanisms are unclear. Our recent findings using monolayers of intestinal cells suggest that the mechanism could be oxidant induced damage to cytoskeletal proteins. However, oxidants and oxidative damage have not been well characterised in IBD mucosa.
Aims: To determine whether there are increases in oxidants and in tissue and cytoskeletal protein oxidation in IBD mucosa.
Methods: We measured nitric oxide (NO) and markers of oxidative injury (carbonylation and nitrotyrosination) to tissue and cytoskeletal proteins in colonic mucosa from IBD patients (ulcerative colitis, Crohn’s disease, specific colitis) and controls. Outcomes were correlated with IBD severity score.
Results: Inflamed mucosa showed the greatest increases in oxidants and oxidative damage. Smaller but still significant increases were seen in normal appearing mucosa of patients with active and inactive IBD. Tissue NO levels correlated with oxidative damage. Actin was markedly (>50%) carbonylated and nitrated in inflamed tissues of active IBD, less so in normal appearing tissues. Tubulin carbonylation occurred in parallel; tubulin nitration was not observed. NO and all measures of oxidative damage in tissue and cytoskeletal proteins in the mucosa correlated with IBD severity. Disruption of the actin cytoarchitecture was primarily within the epithelial cells and paracellular area.
Conclusions: Oxidant levels increase in IBD along with oxidation of tissue and cytoskeletal proteins. Oxidative injury correlated with disease severity but is also present in substantial amounts in normal appearing mucosa of IBD patients, suggesting that oxidative injury does not necessarily lead to tissue injury and is not entirely a consequence of tissue injury. Marked actin oxidation (>50%)—which appears to result from cumulative oxidative damage—was only seen in inflamed mucosa, suggesting that oxidant induced cytoskeletal disruption is required for tissue injury, mucosal disruption, and IBD flare up.
- inflammatory bowel disease
- ulcerative colitis
- Crohn’s disease
- free radicals
- intestinal barrier
- cytoskeleton
- nitric oxide
- oxidative stress
- inflammation
- ROM, reactive oxygen metabolite
- RNM, reactive nitrogen metabolite
- NO, nitric oxide
- iNOS, inducible NO synthase
- IBD, inflammatory bowel disease
- UC, ulcerative colitis
- CD, Crohn’s disease
- UCAI, ulcerative colitis disease activity index
- CDAI, Crohn’s disease activity index
- PVDF, polyvinylidene difluoride
- TBS, Tris buffered saline
- ECL, enhanced chemiluminescence
- OD, optical density
- DNPH, 2,4, dinitrophenylhydrazine
- BSA, bovine serum albumin
- PBS, phosphate buffered saline
- HRP, horseradish peroxidase
- LSCM, laser scanning confocal microscope
Statistics from Altmetric.com
- inflammatory bowel disease
- ulcerative colitis
- Crohn’s disease
- free radicals
- intestinal barrier
- cytoskeleton
- nitric oxide
- oxidative stress
- inflammation
- ROM, reactive oxygen metabolite
- RNM, reactive nitrogen metabolite
- NO, nitric oxide
- iNOS, inducible NO synthase
- IBD, inflammatory bowel disease
- UC, ulcerative colitis
- CD, Crohn’s disease
- UCAI, ulcerative colitis disease activity index
- CDAI, Crohn’s disease activity index
- PVDF, polyvinylidene difluoride
- TBS, Tris buffered saline
- ECL, enhanced chemiluminescence
- OD, optical density
- DNPH, 2,4, dinitrophenylhydrazine
- BSA, bovine serum albumin
- PBS, phosphate buffered saline
- HRP, horseradish peroxidase
- LSCM, laser scanning confocal microscope
The manifestations of inflammatory bowel disease (IBD), including ulcerative colitis (UC) and Crohn’s Disease (CD), wax and wane between active (symptomatic) and inactive (asymptomatic) phases. Although the trigger for the acute attack is not known, the pathophysiological mechanism of tissue damage in the active phase of IBD has been investigated.1,2 It is now widely held that tissue damage in IBD is a result of abnormal mucosal immune reactions to bacterial products and other lumenal factors, reactions that initiate an inflammatory cascade. The inflammatory cascade begins by infiltration of inflammatory cells into the mucosa and release of proinflammatory mediators such as reactive oxygen metabolites (ROM) and reactive nitrogen metabolites (RNM).1–5 These mediators cause tissue damage and result in additional recruitment of inflammatory cells—a vicious cycle that sustains the inflammatory cascade. In this view, a disrupted intestinal barrier both initiates and perpetuates the cascade by exposing luminal factors to the mucosal immune system.6,7 When this vicious cycle is aborted, tissue damage can be repaired. Left unchecked, inflammation is sustained, resulting in intestinal tissue damage and symptoms of the active phase of IBD. Accordingly, it is now critical to identify the most important proinflammatory factors that maintain this vicious cycle and shift the disease from inactive to active phases. Such knowledge could have significant diagnostic, prognostic, and therapeutic impact.
ROM and RNM represent one critical group of proinflammatory factors that could maintain the vicious cycle of IBD.1–5 These oxidants can overwhelm antioxidant defences and, through protein oxidation, DNA strand breaks, and ATP depletion, cause tissue damage. Indeed, several studies have implicated nitric oxide (NO) and other oxidants in the active phase of IBD.5,8–10 We have shown, using monolayers of human colonic cells in culture, that ROM and RNM can oxidise actin and tubulin and that the ensuing cytoskeletal disruption causes barrier dysfunction.11–16 Similar observations have been made in vivo in rats.17,18 However, direct measurements in mucosa of IBD patients showing increases in levels of these oxidants and/or the damaging effects of these oxidants are lacking. Accordingly, the aim of the current study was to measure mucosal NO levels and oxidation of tissue proteins, including cytoskeletal proteins, in patients with active and inactive IBD, and correlate these markers of tissue oxidative stress with indices of disease activity. These data could provide support for the hypothesis that elevated mucosal oxidant levels initiate IBD flare up through oxidative injury to the cytoskeleton, intestinal barrier disruption, and mucosal damage.
METHODS
Subjects
Forty seven IBD patients (20 female; 27 male; mean age 42 years) who underwent colonoscopic examination as part of their clinical evaluation were randomly selected. The diagnosis of IBD was established on the basis of classic clinical, endoscopic, and histological criteria. This group included 22 patients with UC, 11 with CD, and 14 with specific colitis (10 with radiation proctitis; four with diverticulitis). All 11 CD patients had ileocolonic inflammation. For both UC and CD, patients were considered either active (n=15 for UC; n=6 for CD) or inactive (n=7 for UC; n=5 for CD) on the basis of disease activity indexes for UC19,20 or CD.21 All patients with inactive IBD were asymptomatic and had no evidence of mucosal ulceration or friability on endoscopic examination. The majority (32 of 47) of IBD patients were taking IBD related medications. Medications included prednisone (six UC; five CD), mesalamine (13 UC; five CD), and the immunosuppressive medications 6-mercaptopurine or azothioprine (four UC; two CD). The remaining 15 (nine UC; six CD) were taking no IBD related medication. The results were compared with data obtained from 10 subjects who underwent colonoscopy for evaluation of occult blood positive stool or abdominal pain, all of whom proved to have a normal colonoscopic examination (control group).
Each subject underwent colonoscopic examination after a colon preparation with Golytely solution and with conscious sedation (Versed and Demerol). Mucosal biopsy specimens were collected, snap frozen in liquid nitrogen, and stored in a −70°C freezer. In patients with active left sided UC (n=6), biopsies were taken from both inflamed and non-inflamed areas of the mucosa. In patients with active CD, biopsies were only taken from grossly inflamed areas because the patchy pattern of the involvement made it difficult to accurately sample non-involved areas. This study was approved by the Institutional Review Board of Rush Presbyterian Medical Center and was performed after obtaining written consent from subjects.
Chemiluminescence analysis of nitric oxide (NO) concentrations in tissue
NO production was assessed by chemiluminescence.11 Briefly, tissues were homogenised and sonicated, and endogenous nitrate (NO3−) and nitrite (NO2−), the metabolic degradation products of NO, were reduced to NO using vanadium (III) (Sigma, St Louis, Missouri, USA) and HCl at 90°C. Chemiluminescence was measured using a Sievers NOA 280 analyser (Sievers, Colorado, USA). NO was expressed in μmol/g protein and calculated by comparison with the chemiluminescence of a standard solution of NaNO2.
Slot immunoblotting measurement of mucosal protein oxidation and protein nitration
Oxidation and nitration of mucosal proteins were assessed by measuring protein carbonyl and protein nitrotyrosine formation using a slot blotting method described previously.22 Biopsied samples were homogenised and protein concentrations were assessed by the Bradford method.23 Equal amounts of total protein (5 μg) were blotted to polyvinylidene difluoride (PVDF) membranes.
To determine carbonyl immunoreactivity, membranes were blocked in 5% non-fat milk in Tris buffered saline (TBS) at room temperature for one hour, incubated with a monoclonal rabbit anti-carbonyl antibody (1:25 000; Upstate Biotech, Lake Placid, New York, USA) in blocking solution at 4°C overnight, washed 5× for five minutes each wash (in 1% non-fat milk, 0.1% Tween 20 TBS), incubated with monoclonal peroxidase conjugated goat anti-rabbit antibody (1:5000) in blocking buffer at room temperature for one hour, and washed again 5×. Membranes were soaked in enhanced chemiluminescence (ECL) reagents and exposed to ECL hyperfilm.
A similar procedure was used to determine nitrotyrosine immunoreactivity except that a monoclonal mouse anti-nitrotyrosine antibody (1:5000) was used. We then quantified the relative levels of oxidised proteins by measuring the optical density (OD) of the bands corresponding to anti-carbonyl or anti-nitrotyrosine immunoreactivity with a laser densitometer. Carbonyl or nitrotyrosine formation was expressed as the ratio of carbonyl or nitrotyrosine formation in the treatment group divided by carbonyl or nitrotyrosine formation in the corresponding carbonylated or nitrated (tissue) standard run concurrently.
Western immunoblotting measurement of carbonylation and nitration of actin and tubulin cytoskeletons
Oxidation and nitration of actin and tubulin were assessed by measuring carbonyl and nitrotyrosine formation as described previously.11,12 Briefly, tissues were homogenised and processed for polyacrylamide gel electrophoresis fractionation and western immunoblotting. The identity of the bands was confirmed as actin or tubulin by comparison with standards run oncurrently. In separate blots, specific monoclonal anti-actin or anti-tubulin antibody further confirmed the identity of actin or tubulin. To avoid oxidation during sample processing, all buffers contained 0.5 mM dithiothreitol and 20 mM 4,5-dihydroxy-1,3-benzene sulphonic acid (Sigma).
Samples were blotted to a PVDF membrane followed by successive incubations in 2 N HCl and 2,4, dinitrophenylhydrazine (DNPH 100 μg/ml in 2 N HCl; Sigma) for five minutes each. Membranes were washed 3× in 2 N HCl and washed 7× in 100% methanol (five minutes each), followed by blocking for one hour in 5% bovine serum albumin (BSA) in 10× phosphate buffered saline (PBS)/Tween 20 (PBS-T). Membranes were then incubated for one hour in 1% BSA/PBS-T buffer containing anti-DNPH (1:25 000 dilution) (Molecular Probes, Eugene, Oregon, USA) and further incubated with a horseradish peroxidase (HRP) conjugated secondary antibody (1:4000 dilution, one hour) (Molecular Probes).
To determine the nitrotyrosine content of actin or tubulin we used a similar method, except following the blocking step above (that is, BSA/PBS-T buffer), membranes were incubated with 2 μg/ml monoclonal anti-nitrotyrosine antibody for one hour (Upstate Biotech) followed by the HRP conjugated secondary antibody as above. Wash steps and film exposure were as in a standard western blot protocol. Relative levels of oxidised or nitrated actin or tubulin were then quantified by measuring, with a laser densitometer, the OD of the bands corresponding to anti-DNP or anti-nitrotyrosine immunoreactivity. Comparing OD values, immunoreactivity was expressed as the percentage of carbonyl or nitrotyrosine formation in the treatment group compared with that in the maximally oxidised or nitrated actin or tubulin standard run concurrently.
Staining of the F-actin cytoskeleton in the mucosa of patients with IBD
To study morphological perturbations in the cytoskeleton, fluorescent staining was used.13,18 Briefly, mucosal tissues were fixed and subsequently processed for incubation with FITC-phalloidin (specific for F-actin; Sigma), 1:33 dilution for one hour at 37°C. Slides were washed thrice in D-PBS, once with deionised H2O, and subsequently mounted in aquamount. Following staining, mucosal tissues were observed with an argon laser (λ=488 nm) using a 63× oil immersion plan apochromat objective, NA 1.4 (Zeiss, Germany). Desired areas of mucosa were processed using the image processing oftware on a Zeiss ultra high resolution laser scanning confocal micscope (LSCM). The cytoskeletal filaments were examined in a blinded fashion for their overall morphology, orientation, and disruption as we previously described.13,18 Each slide was examined in eight different fields by LSCM and F-actin was then carefully assessed for fragmentation, kinking, disruption, and collapse. Slides were decoded only after examination was complete.
Statistical analysis
Data are presented as mean (SEM). All experiments were carried out with a sample size of 5–15 observations per group. Statistical analyses between or among groups were carried out using analysis of variance followed by Tukey’s post hoc test. Correlation analyses were done using the Pearson test for parametric analysis or, when applicable, the Spearman test for non-parametric analysis. For certain correlations, we used UC disease activity index (UCAI)19,20 or CD activity index (CDAI).21 To provide a unifying score for both UC and CD, we also calculated an “IBD severity score” that ranged from 1 to 4 and was based on the following: 1=inactive disease; 2=mildly active disease (CD: CDAI =150–200; UCAI=2–5); 3=moderately active (CD: CDAI=201–300, UCAI=6–9); 4=severely active (CD: CDAI >300, UCAI >9). A p value <0.05 was considered statistically significant.
RESULTS
Mucosal NO levels in IBD patients
Low levels of NO were present in control colonic tissues (fig 1A). NO levels were elevated in all patients with active IBD or specific colitis. NO levels were substantially higher in active IBD than in inactive IBD. NO levels in normal appearing non-inflamed mucosa (in the proximal sigmoid) of patients with left sided active UC were still greater than in controls. In active UC, ulcerated mucosa had higher NO levels than normal appearing mucosa and but this difference was not significant (paired analysis). NO levels were higher in inflamed mucosa from active CD than in active UC (NS). NO levels correlated significantly with IBD severity score (fig 1B).
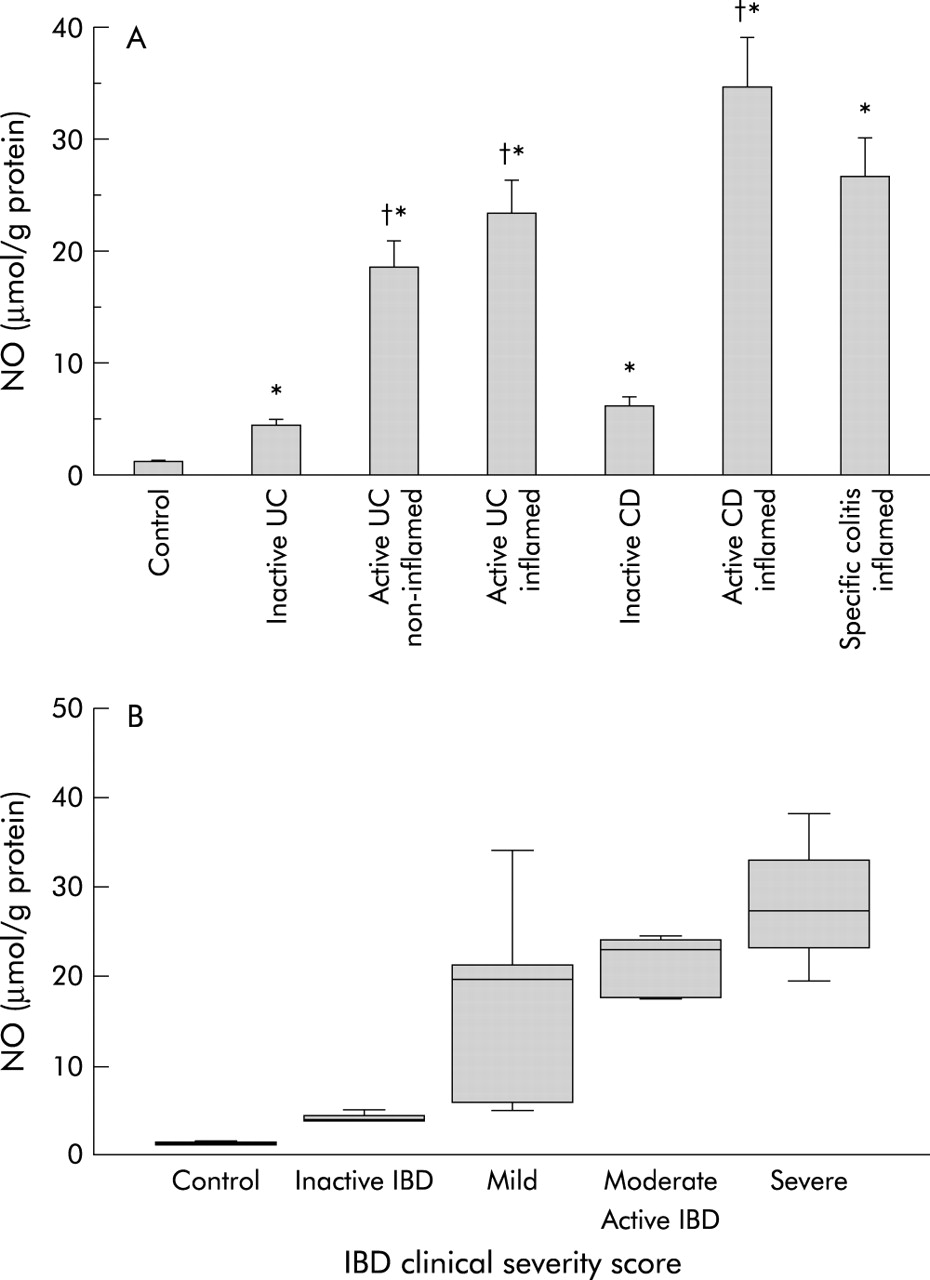
(A) Concentrations of nitric oxide (NO) in homogenates from intestinal mucosal biopsies from: controls (n=10); patients with inactive ulcerative colitis (UC) (n=7); normal appearing non-inflamed mucosa of patients with active UC (n=6); inflamed ulcerated mucosa of patients with active UC (n=15); inactive Crohn’s disease (CD) (n=5); active CD (n=6); and inflamed mucosa of patients with specific colitis (n=14). NO levels were assessed by chemiluminescence. Data are mean (SEM); *p<0.05 versus controls, †p<0.05 versus inactive inflammatory bowel disease (IBD). (B) Correlation between mucosal NO levels and IBD disease severity score. Correlations are from: controls (n=5); patients with inactive IBD (UC=7, CD=5); and active IBD (UC=14, CD=5) (non-parametric correlation, Spearman correlation coefficient 0.81; p<0.0001).
Mucosal levels of nitrotyrosine in IBD patients
Mucosal nitration (nitrotyrosine immunoreactivity) was also elevated in patients with IBD regardless of disease type or disease activity (fig 2A, 2B) with (a) active IBD (UC and CD) being higher than inactive IBD, (b) active IBD being higher than active specific colitis, and (c) active CD similar to active UC. Unlike NO levels, nitrotyrosine levels in normal appearing mucosa from patients with active left sided UC were significantly higher than in inflamed mucosa from the same patients (paired analysis). There were positive correlations between: (a) NO and nitrotyrosine (fig 2C) and (b) nitrotyrosine and disease severity score (fig 2D).

(A) Slot immunoblotting analysis of levels of anti-nitrotyrosine (nitration) immunoreactivity of proteins in mucosal pinch biopsies. (B) Representative slot immunoblot of the nitrated mucosal proteins. Mucosal homogenates were analysed by slot blotting and processed for autoradiography and densitometry. Nitration immunoreactivity was expressed as follows: nitrotyrosine formation (optical density) in the patient group divided by the nitrated tissue standards, expressed as a percentage. Controls (n=10); patients with inactive ulcerative colitis (UC) (n=7); normal appearing non-inflamed mucosa of patients with active UC (n=6); inflamed ulcerated mucosa of patients with active UC (n=15); inactive Crohn’s disease (CD) (n=5); active CD (n=6); and inflamed mucosa of patients with specific colitis (n=14). *p<0.05 versus controls, †p<0.05 versus inactive inflammatory bowel disease (IBD), ‡p<0.05 versus non-inflamed UC. (C) Correlation between mucosal nitrotyrosine levels and nitric oxide (NO) (r=0.65, r2=0.43, p<0.0001). (D) Correlation between mucosal nitrotyrosine and IBD disease severity score (non-parametric correlation, Spearman correlation coefficient 0.84; p<0.0001). Number of subjects used for analysis as in previous figures.
Mucosal levels of carbonyls in IBD patients
Carbonylation, like nitration, was higher in all patients with IBD regardless of disease type (fig 3A, 3B). Carbonylation was higher in active CD and UC than in inactive CD and UC, respectively. Carbonylation was higher in inflamed mucosa than in non-inflamed mucosa of the same patients with active UC (paired analysis). There were no significant differences between carbonyl levels in the inflamed mucosa of patients with active UC compared with active CD or active specific colitis.
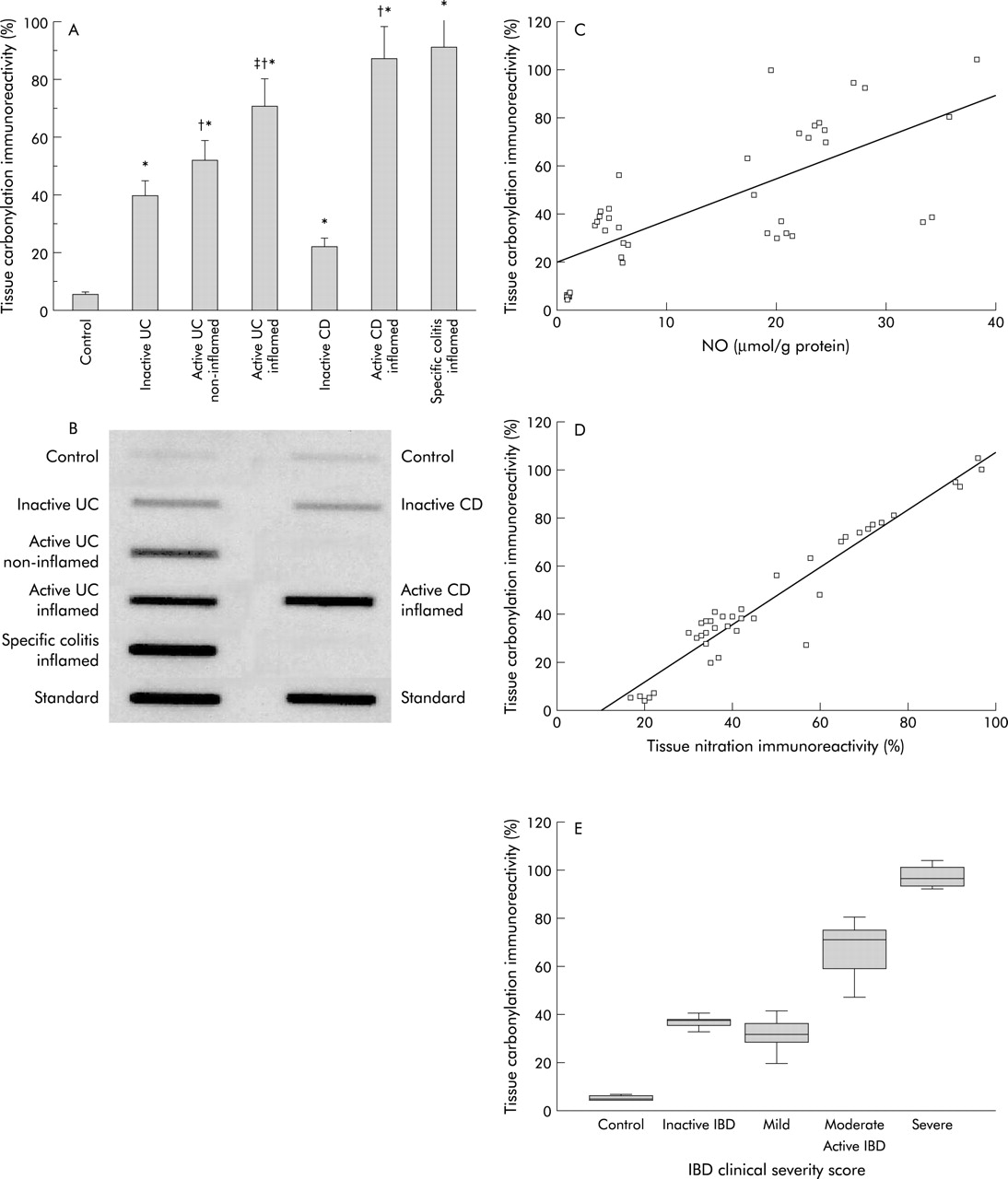
(A) Slot immunoblotting analysis of leves of carbonylation (anti-dinitrophenylhydrazone) immunoreactivity of proteins in mucosal pinch biopsies. (B) Representative slot blot of the carbonylated tissue proteins. Oxidation was expressed as carbonyl formation (that is, optical density) in the patient group divided by the oxidised tissue standards, expressed as a percentage. From controls (n=10); patients with inactive ulcerative colitis (UC) (n=7); normal appearing non-inflamed mucosa of patients with active UC (n=6); inflamed ulcerated mucosa of patients with active UC (n=15); inactive Crohn’s disease (CD) (n=5); active CD (n=6); and inflamed mucosa of patients with specific colitis (n=14). *p<0.05 versus controls, †p<0.05 versus inactive inflammatory bowel disease (IBD), ‡p<0.05 versus non-inflamed UC. Correlation between mucosal carbonylation levels and mucosal nitric oxide (NO) levels (r=0.72, r2=0.51, p<0.0001) (C), between mucosal carbonylation levels and nitrotyrosine levels (r=0.96, r2=0.93, p<0.0001) (D), and between mucosal carbonylation levels and IBD disease severity score (non-parametric correlation, Spearman correlation coefficient 0.81; p<0.0001) (E). Number of subjects used for analysis was as above.
Similar to nitration, positive correlations were observed between: (a) carbonylation and NO levels (fig 3C), (b) carbonylation and nitrotyrosine levels (fig 3D), and (c) carbonylation and disease severity score (fig 3E).
Mucosal levels of oxidised and nitrated actin and tubulin
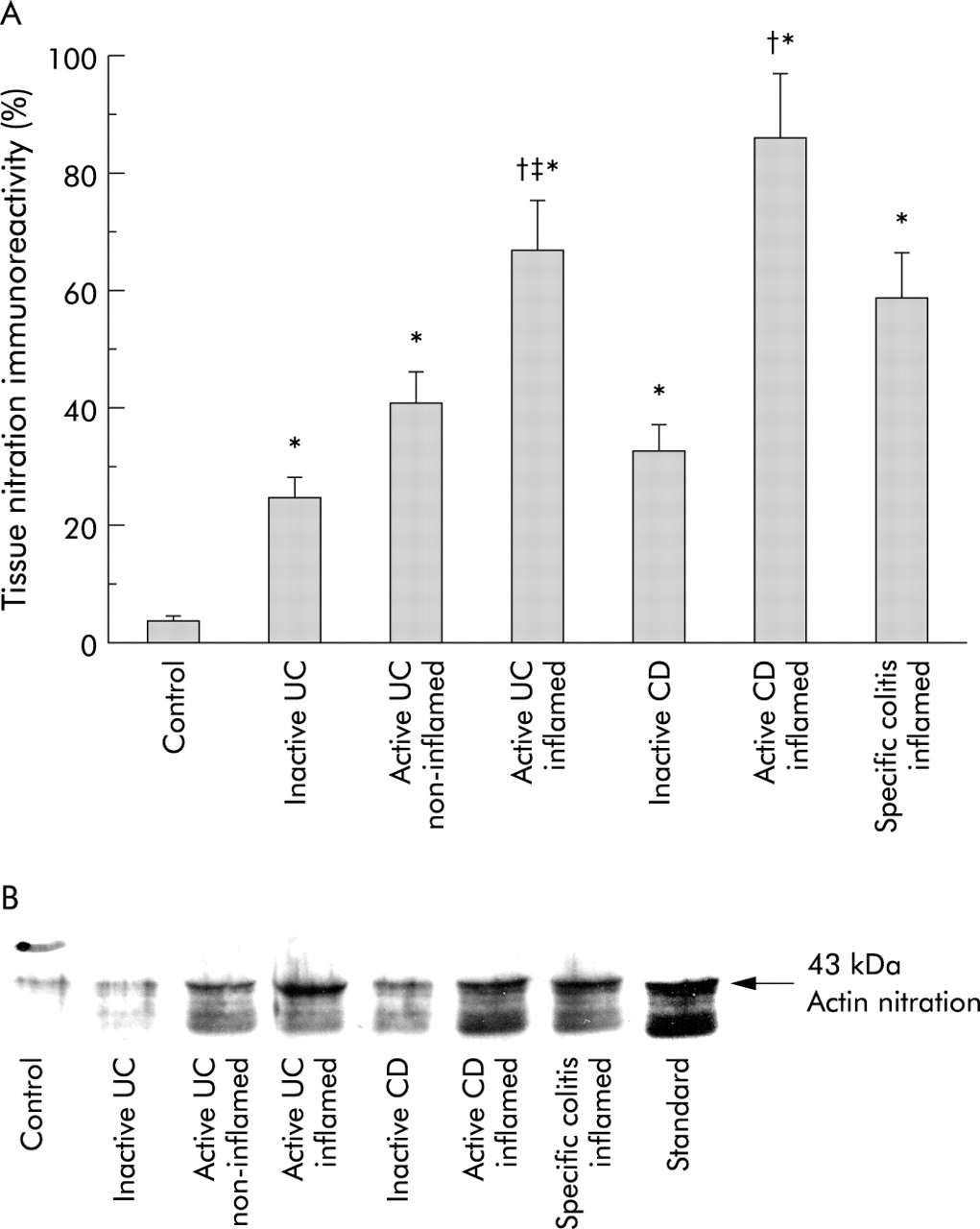
Both carbonylation (fig 4A, 4B) and nitration (fig 5A, 5B) of actin (43 kDa) were increased in the mucosa of patients with colitis, regardless of type or activity. Actin nitration and carbonylation were significantly less in the non-inflamed mucosa of patients with active UC than in inflamed mucosa from the same patient (paired analysis). Both actin nitration and carbonylation were higher in active CD and UC than in the corresponding inactive disease. Figures 4B and 5B show that the identity of the bands was confirmed to be oxidised actin using known carbonylated and nitrated actin standards.

(A) Immunoblotting analysis of carbonylation (anti-dinitrophenylhydrazone (DNP)) immunoreactivity of the actin cytoskeleton from intestinal mucosa. A representative blot for actin carbonylation is shown in (B). Western blots of mucosal homogenates were processed for actin fractionation sodium dodecyl sulphate-polyacrylamide gel electrophoresis and processed sequentially using monoclonal anti-DNP and horseradish peroxidase conjugated secondary antibodies. The region of gel shown is between the M 42 000 and 63 000 pre-stained molecular weight standards, which were run in adjacent lanes. Carbonlyation of actin was expressed as carbonyl formation (that is, optical density) in the appropriate group divided by the oxidised actin standard. Controls (n=10); patients with inactive ulcerative colitis (UC) (n=7); normal appearing non-inflamed mucosa of patients with active UC (n=6); inflamed ulcerated mucosa of patients with active UC (n=15); inactive Crohn’s disease (CD) (n=5); active CD (n=6); and inflamed mucosa of patients with specific colitis (n=14). *p<0.05 versus controls, †p<0.05 versus inactive inflammatory bowel disease, ‡p<0.05 versus non-inflamed UC.

(A) Immunoblotting analysis of anti-nitrotyrosine immunoreactivity of actin from the intestinal mucosa and a representative immunoblot of this actin nitration (B). Nitration of actin was expressed as nitrotyrosine formation (optical density) in the patient group divided by the nitrated actin standard. Western blots were processed as in fig 4 except that monoclonal anti-nitrotyrosine was used as the primary antibody. Representative blot from controls (n=10); patients with inactive ulcerative colitis (UC) (n=7); normal appearing non-inflamed mucosa of patients with active UC (n=6); inflamed ulcerated mucosa of patients with active UC (n=15); inactive Crohn’s disease (CD) (n=5); active CD (n=6); and inflamed mucosa of patients with specific colitis (n=14). *p<0.05 versus controls, †p<0.05 versus inactive inflammatory bowel disease, ‡p<0.05 versus non-inflamed UC.
Similar to actin, tubulin (50 kDa) was carbonylated more in the colonic mucosa of patients with colitis (fig 6A, 6B). Carbonyl levels were less in inactive disease than in inflamed mucosa of active disease. Tubulin carbonylation in non-inflamed mucosa was significantly less than in inflamed mucosa in the same active UC patient (paired analysis). There was no significant difference between tubulin carbonylation and actin carbonylation. There was no detectable nitration of tubulin (fig 7). The identity of tubulin was confirmed using oxidised and nitrated tubulin standards (figs 6B, 7).

(A) Quantitative immunoblotting analysis of anti-dinitrophenylhydrazone immunoreactivity of the tubulin cytoskeleton from mucosal biopsies. A representative blot showing the oxidation of tubulin is shown in (B). Tubulin fractions from mucosal homogenates were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis and analysed by autoradiography and densitometry. The region of gel shown is between the M 50 000 and 70 000 pre-stained molecular weight standards, which were run in adjacent lanes. Representatives blot from controls (n=10); patients with inactive ulcerative colitis (UC) (n=7); normal appearing non-inflamed mucosa of patients with active UC (n=6); inflamed ulcerated mucosa of patients with active UC (n=15); inactive Crohn’s disease (CD) (n=5); active CD (n=6); and inflamed mucosa of patients with specific colitis (n=14). *p<0.05 versus controls, †p<0.05 versus inactive inflammatory bowel disease, ‡p<0.05 versus non-inflamed UC.

Representative immunoblotting of anti-nitrotyrosine immunoreactivity of the tubulin fraction from mucosal biopsies. Note the absence of tubulin nitration in any group (same groups as in fig 6B). Tubulin fractions from mucosal homogenates were processed for sodium dodecyl sulphate-polyacrylamide gel electrophoresis and immunoblotted using monoclonal anti-nitrotyrosine and horseradish peroxidase conjugated secondary antibodies. The region of gel shown is between the M 50 000 and 70 000 pre-stained molecular weights, which were run in adjacent lanes. Representative blot from controls (n=10); patients with inactive ulcerative colitis (UC) (n=7); normal appearing non-inflamed mucosa of patients with active UC (n=6); inflamed ulcerated mucosa of patients with active UC (n=15); inactive Crohn’s disease (CD) (n=5); active CD (n=6); and inflamed mucosa of patients with specific colitis (n=14).
Figure 8B shows that the F-actin cytoskeleton in the epithelial cells of the colonic mucosa of patients with active IBD is disrupted. F-actin cytoarchitecture was fragmented, disorganised, and collapsed (arrows). In contrast, normal mucosa (fig 8A) exhibited an intact and smooth distribution of F-actin.

Fluorescent staining of F-actin cytoskeleton as captured by ultra high resolution laser scanning confocal microscopy revealing its intracellular distribution in intestinal mucosa from a patient with inflammatory bowel disease (IBD) and a normal control. Normal mucosa (A) exhibits an intact, continuous, and smooth distribution of F-actin. In contrast, the F-actin in the colonic mucosa of a patient with active ulcerative colitis (UC) (B) appears to be fragmented, disorganised, and collapsed (arrows). Bar=100 μm. Representative photos from n=4 IBD (three active UC, one active Crohn’s disease) and three controls.
DISCUSSION
A growing body of evidence indicates that oxidants, including free radicals, play a key role in the pathophysiology of tissue injury in IBD, particularly in the initiation and perpetuation of inflammation and in the subsequent tissue damage. Firstly, we2,24,25,26 and others1,4,5,8 reported increases in oxidants in IBD tissues and in experimental colitis. Secondly, antioxidants prevent tissue damage in experimental colitis.26,27 Indeed, many drugs used in the treatment of colitis, such as mesalamine, are antioxidants.28,29 Nevertheless, mechanisms underlying oxidant induced tissue damage have not been established.
For example, neither the oxidants nor their protein or organelle targets are well characterised. Initially, ROM, in particular superoxide anion, were believed to be the critical oxidants in IBD. However, it subsequently became clear that superoxide anion is a rather weak oxidant in vivo and that other neutrophil derived ROM such as H2O2, HOCl, and hydroxyl radicals were more injurious.30 Indeed, oxidative damage in inflamed IBD mucosa appears to be due to high levels of these neutrophil derived oxidants1–4 and to low tissue levels of antioxidants such as catalase and glutathione.31
Moreover, RNM such as NO may play an equally important or even bigger role in tissue injury32–35: (i) high NO levels, which result from inducible NO synthase (iNOS) activation, are associated with tissue damage10,32,34–36; (ii) iNOS is present in inflammatory cells such as neutrophils and macrophages and in intestinal epithelial and endothelial cells; (iii) iNOS is activated in the presence of proinflammatory factors such as cytokines—a milieu commonly found in active IBD; (iv) high iNOS activity has been found in experimental models of colitis and in the intestinal mucosa of patients with IBD8–10; and (v) high levels of the end products of NO metabolism, nitrates and nitrites, were found in the plasma, stool, and colonic lumen of IBD patients.37–39 These indirect measurements may not accurately reflect changes in NO levels in intestinal mucosa. Bacteria and diet might have contributed to elevated NO levels. Lacking has been direct measurements in IBD mucosa of oxidants and oxidative damage to specific tissue proteins and organelles.
Herein, we begin to fill that gap by reporting that NO is increased in inflamed colonic mucosa of patients with active IBD. This was also observed in patients with specific colitis and is thus not specific for IBD (UC and CD). Because NO levels were also elevated, albeit less so, in normal appearing mucosa of patients with inactive and even active IBD, other factors must contribute to tissue damage either by potentiating the injuriousness of NO or by blunting tissue antioxidant activity. Consistent with these results is a finding in collagenous colitis where there was a high level of lumenal NO in patients who had, macroscopically, normal appearing mucosa.40
Our findings are consistent with earlier reports10,12,13,35 that suggest that the deleterious effects of NO and other RNM are due to their ability to nitrate critical proteins, forming nitrotyrosine. Indeed, elevations in mucosal NO levels correlated with elevations in nitrotyrosine (adjusted r2=0.43), suggesting that O is the major source of the increased protein nitration. Surprisingly, mucosal NO levels in inactive CD were markedly less than in active CD and yet their nitrotyrosine levels were comparable. One explanation is that other RNM contribute to tissue nitration in CD. Or, there could be a ceiling effect for NO induced tissue nitration whereby relatively low levels of NO can cause near maximal nitration. Similar to NO levels, high levels of nitrotyrosine are not specific for IBD as nitrotyrosine was also increased in specific colitis.
Protein carbonylation, another form of oxidative damage,13,22 was also increased in colitis. Like nitration, carbonylation was increased even in the non-inflamed mucosa of patients with active and inactive IBD but less so than for inflamed tissue. This suggests that colonic mucosa can tolerate “subthreshold” levels of protein nitration and carbonylation which is insufficient to disrupt the cytoarchitecture and cause tissue injury. Indeed, our findings are consistent with this idea that gross tissue injury occurs in IBD only after oxidative stress reaches a critical level required to damage crucial proteins and that actin and tubulin are two such crucial proteins. Our findings of large increases in actin and tubulin oxidation in active inflamed IBD mucosa and smaller increases in normal appearing mucosa in patients with active or inactive IBD are consistent with this idea. This conclusion is based in part on our previous demonstration that depolymerisation and disruption of the actin and tubulin cytoskeletons in monolayers of intestinal cells occur only when oxidative stress (carbonylation and nitration of actin and tubulin) is greater than about 55%.11–15 As noted, cytoskeletal disruption in monolayers was associated with barrier disruption. Our current study suggests that this “threshold” is also present in IBD mucosa and is also about 55% (fig 9).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Analysis of oxidative damage versus nitric oxide (NO) levels from the sigmoid mucosa of patients with ulcerative colitis (UC). Note the parallel increases in several measures of oxidative damage (for example, tissue nitration and carbonylation, actin nitration and carbonylation, and tubulin carbonylation) and NO overproduction. Measures of oxidative damage are lower than 55% in normal appearing mucosa of patients with inactive UC and in the non-inflamed mucosa of patients with active left sided UC. In contrast, inflamed ulcerated mucosa of patients with active disease show increased oxidative damage well above the 55% levels.
Our prior studies on monolayers showed that oxidation and/or nitration to either actin or tubulin can cause barrier disruption13,14 and that both of these cytoskeleton filaments might be involved in oxidant induced disruption of the intestinal barrier. Our current findings suggest that colonic mucosa can tolerate higher levels of protein nitration than protein carbonylation and that oxidative damage to actin is probably more critical than tubulin for tissue injury in IBD. However, the current study cannot directly help us determine the relative importance of actin versus tubulin damage or the relative importance of oxidation versus nitration. Our current study however suggests that tissue damage may occur in the absence of tubulin nitration.
The fact that a substantial proportion of maximal oxidative tissue damage was already present in normal appearing mucosa in patients with both active and inactive IBD strongly suggests that a large fraction of this oxidative injury is not secondary to tissue injury. Thus it is possible that oxidative injury in patients with inactive IBD makes their mucosa susceptible to additional injurious factors that can increase tissue disruption, initiate an inflammatory cascade (vicious cycle/ positive feed back loop), and cause IBD flare up.
A possible mechanism through which oxidant induced cellular injury can initiate an inflammatory cascade that can lead to or exacerbate tissue damage in IBD is barrier disruption. Decreased intestinal barrier integrity, which has been demonstrated in patients with IBD,6,7 is now believed to be an integral factor in both the initiation and perpetuation of the inflammatory cascade in IBD.6 Using monolayers of intestinal cells, we11–16 and others41–45 showed that: (1) an intact cytoskeleton, especially the actin and tubulin components, is essential to barrier integrity; (2) oxidants can disrupt barrier function of intestinal monolayers in vitro11–16; (3) oxidant induced loss of barrier integrity requires oxidative damage to tubulin and actin; (4) complete disruption of either microtubules (by colchicine) or the actin cytoskeleton (by cytochalasin D) or partial disruption of both (by oxidants) results in barrier disruption; and (5) protection of the intestinal barrier against oxidant induced damage can be achieved by stabilisation of either the actin cytoskeleton or the tubulin cytoskeleton. We now provide evidence that oxidant induced cytoskeletal disruption is important in tissue damage in patients with IBD. We showed that oxidation of actin is associated with marked disruption of the actin cytoskeleton in the mucosa of patients with active IBD. Hence increased actin and tubulin oxidation and cytoskeletal disruption in the colonic mucosa of IBD patients could conceivably cause intestinal barrier disruption. In this view, barrier disruption then allows penetration of proinflammatory factors and initiates the sustained inflammatory cascade that is required for mucosal injury and IBD flare up. Further studies are needed to see if actin and tubulin oxidation correlate with intestinal barrier disruption and the onset of IBD flare up.
In summary, our study indicates that increased levels of tissue oxidation occur in the mucosa of patients with all types of colitis studied but marked cytoskeletal protein oxidation and cytoskeletal disarray occurs in injured mucosa of patients with active colitis. Based on these findings, we propose a model in which barrier disruption occurs only when oxidation exceeds a threshold value and causes cytoskeletal disarray. Inflammation can then be initiated, which causes additional oxidative damage, additional barrier disruption, and sustained inflammation (vicious cycle). When enough mucosal damage accumulates, there is a shift in disease state from inactive to active IBD (flare up). This model suggests several different strategies for identifying new therapeutic targets and developing new therapeutic agents that can intervene in and slow, stop, or prevent one or more of the steps in what appears to be the final common pathway of IBD pathophysiology.
Acknowledgments
This work was supported in part by a grant from Rush University Medical Center and the American College of Gastroenterology (ACG). Portions of this work were presented in abstract form at the annual meeting of the American Gastroenterological Association in Atlanta, GA, 2001.