Article Text

Abstract
Background: We previously reported that gastrin induces expression of CXC chemokines through activation of nuclear factor κB (NFκB) in gastric epithelial cells that express gastrin receptor.
Aims: To clarify gastrin receptor mediated signals leading to activation of NFκB.
Methods: MKGR26 cells were created by transfecting gastrin receptor cDNA into MKN-28 cells. Degradation of inhibitor κB (IκB) and phosphorylation of protein kinase C (PKC)-δ were both detected by western blot analysis. NFκB activation was determined by luciferase assay and electrophoretic mobility shift analysis.
Results: Gastrin induced degradation of IκB-α and activation of NFκB, which was abolished by the selective gastrin receptor antagonist L-740,093 and the general PKC inhibitor GF109203X. Gastrin induced phosphorylation of PKC-δ, and its inhibitor rottlerin partially suppressed NFκB activation. However, the mitogen activated protein kinase (MAPK) kinase inhibitor PD98059, p38 MAPK inhibitor SB203580, and tyrphostin AG1478 had no effect on NFκB activation. Introduction of the dominant negative mutant of IκB kinase, of NFκB inducing kinase, and of tumour necrosis factor receptor associated factor 6 (TRAF6), but not that of TRAF2, inhibited gastrin induced activation of NFκB.
Conclusions: Gastrin activates NFκB via a PKC dependent pathway which involves IκB kinase, NFκB inducing kinase, and TRAF6.
- nuclear factor κB
- inhibitor κB
- gastrin
- gastrin receptor
- protein kinase C
- tumour necrosis factor receptor associated factor 6
- NFκB, nuclear factor κB
- IκB, inhibitor κB
- MAPK, mitogen activated protein kinase
- PKC, protein kinase C
- EGF, epidermal growth factor
- MEK, MAPK kinase
- IKK, IκB kinase
- NIK, NFκB inducing kinase
- TGF-β, transforming growth factor β
- TAK1, TGF-β activated kinase 1
- TNF, tumour necrosis factor
- TRAF, TNF receptor associated factor
- IL, interleukin
- GPCR, G protein coupled receptor
- EMSA, electrophoretic mobility shift assay
- dn, dominant negative
- FCS, fetal calf serum
- EDTA, ethylenediamine tetraacetic acid
- BSA, bovine serum albumin
Statistics from Altmetric.com
- nuclear factor κB
- inhibitor κB
- gastrin
- gastrin receptor
- protein kinase C
- tumour necrosis factor receptor associated factor 6
- NFκB, nuclear factor κB
- IκB, inhibitor κB
- MAPK, mitogen activated protein kinase
- PKC, protein kinase C
- EGF, epidermal growth factor
- MEK, MAPK kinase
- IKK, IκB kinase
- NIK, NFκB inducing kinase
- TGF-β, transforming growth factor β
- TAK1, TGF-β activated kinase 1
- TNF, tumour necrosis factor
- TRAF, TNF receptor associated factor
- IL, interleukin
- GPCR, G protein coupled receptor
- EMSA, electrophoretic mobility shift assay
- dn, dominant negative
- FCS, fetal calf serum
- EDTA, ethylenediamine tetraacetic acid
- BSA, bovine serum albumin
The peptide hormone gastrin regulates not only acid secretion but also growth and differentiation of gastric epithelial cells.1 In addition, we recently reported that gastrin is capable of inducing expression of interleukin 8 (IL-8), a potent chemotactic and activating factor for leucocytes, in gastric epithelial cells.2 This suggests that gastrin may participate in gastric inflammation under hypergastrinaemic conditions. In general, the transcription factor nuclear factor κB (NFκB) is required for activation of the IL-8 promoter gene by various stimuli.3–,5 We previously demonstrated that NFκB is also critical for gastrin activation of the IL-8 promoter gene.2
The gastrin receptor is a member of the family of G protein coupled receptors (GPCRs) that contain seven membrane spanning regions.6,7 Agonist binding to this receptor activates phospholipase C, leading to hydrolysis of phosphatidylinositol biphosphate, thus generating inositol 1,4,5-triphosphate and diacylglycerol, which mobilise intracellular Ca2+ and activate protein kinase C (PKC), respectively.6,7 Previous studies have shown that gastrin receptor mediated signals involve not only PKC but also other serine/threonine kinases such as phosphatidylinositol 3-kinase, Raf kinase, mitogen activated protein kinase (MAPK) kinase (MEK), and MAPK.7–,9 Furthermore, gastrin transactivates epidermal growth factor (EGF) receptor through induction of EGF-like growth factors.10,11 However, the mechanisms whereby gastrin activates NFκB are largely unknown.
Tumour necrosis factor (TNF) receptor and IL-1 receptor mediated upstream signals leading to activation of NFκB have been well documented in the literature.12–,17 NFκB is composed of homo- and heterodimers of Rel family members which are held in the cytoplasm by the inhibitor κB (IκB) proteins. An IκB kinase (IKK) complex, composed of IKKα and IKKβ, induces phosphorylation of two serine residues of IκB near its N terminus, resulting in ubiquitylation and degradation of IκB by 26S proteasome. After destruction of IκB, NFκB can then translocate to the nucleus thus activating target genes. The IKK complex is activated by NFκB inducing kinase (NIK) and NIK itself is activated via phosphorylation of its Thr559.18,19 TNF receptor associated factor (TRAF) family of adaptor proteins, which are recruited to ligand bound receptors, are involved in the activation of NIK.20 Although the precise mechanistic role of TRAFs in NIK activation is unclear, transforming growth factor β (TGF-β) activated kinase 1 (TAK1) appears to be associated with TRAF6 and activates NIK in an IL-1 signalling pathway leading to activation of NFκB.21,22
The present study was performed to clarify the mechanism of gastrin induced activation of NFκB in gastric epithelial cells. We previously established a gastric epithelial cell line that stably expresses gastrin receptor by DNA transfection.2 Here we report on the effect of inhibitors and/or dominant negative mutants of several protein kinases on gastrin induced activation of NFκB using this model system.
MATERIALS AND METHODS
Reagents
RPMI-1640 medium, fetal calf serum (FCS), and geneticin (G418) were purchased from Gibco BRL (Grand Island, New York, USA). Gastrin 17 was obtained from Peptide Institute (Osaka, Japan). MEK inhibitor PD98059 and PKC inhibitor GF109203X were purchased from Sigma (St Louis, Missouri, USA). Rottlerin, an inhibitor of PKC-δ and PKC-θ, HBDDE, an inhibitor of PKC-α and PKC-γ, Go6976, an inhibitor of PKC-α and PKC-β, and p38 MAPK inhibitor SB203580 were obtained from Calbiochem (San Diego, California, USA). Rabbit antihuman IκB-α antibody and rabbit antiphosphorylated (Thr505) PKC-δ antibody were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, California, USA) and Cell Signaling Technology (Beverly, Massachusetts, USA), respectively. [α-32P]dCTP was purchased from Dupont-NEN (Boston, Massachusetts, USA).
Selective gastrin receptor antagonist L-740,09323 and an EGF receptor specific tyrphostin AG147824 were generously donated by Merck Sharp & Dohme Research Laboratories (Rahway, New Jersey, USA) and Dr A Levitzki (Hebrew University Jerusalem, Israel), respectively.
Cell line
MKGR26 cells were established in our laboratory by transfection of the full length human CCK-B/gastrin receptor cDNA into MKN-28,2 a human gastric cancer cell line.25 The control cell line (MKN-neo cells) was created by transfection of pSV2-neo alone into MKN-28 cells.2 Cells were grown in RPMI-1640 medium supplemented with 10% FCS, 100 units/ml penicillin G, and 100 mg/ml streptomycin.
Plasmids
The pNFκB-LUC containing five copies of consensus NFκB site linked to a minimal E1B promoter-luciferase reporter gene was purchased from Stratagene (La Jolla, California, USA). The expression vectors pRK5-IKKα(K44A), pRK5-IKKβ(K44A), pRK5-NIK(KK429-430AA), pRK5-TRAF2(87-501), and pRK5-TRAF6(289-522) were donated by Tularik Inc.20 pCMV-HA-TAK1(K63W) was a gift from Dr K Matsumoto (Nagoya University, Japan).21 The expression vectors pTB701HA/PKC-δ26 (wild-type PKC-δ) and pHACE-PKC-δ (K/R)27 were donated by Dr Y Ono (Kobe University, Japan) and Dr Jae-Won Soh (Colombia University, New York, USA) respectively. pEF-PKC-θ(K/R)28 was a gift from Dr Gottfried Baier (University of Innsbruck, Austria).
Transfections and luciferase assay
Lipofectamine-Plus (Gibco BRL) was used for transfections. In a typical experiment, for a 35 mm diameter dish containing 1×106 MKGR26 cells, 0.5 μg of pNFκB-LUC to be transfected was used. In each experiment, cells were cotransfected with 0.1 μg of pRL-SV40 (Promega, Madison, Wisconsin, USA) as an internal standard. Twenty four hours after transfection of the luciferase containing reporter genes, gastrin 17 was added to the medium at a concentration of 10−8 M. To determine the transfection efficiency, MKGR26 cells were transfected with pEGFP-C1 containing green fluorescence protein gene (Life Technologies, Grand Island, New York, USA). After 24 hours, the transfected cells were observed in vivo through an inverted laser scanning microscope (Zeiss LSN510). We achieved a transient transfection efficiency of 15 (2)% (mean (SD), n=3) for MKGR26 cells using this protocol.
After 24 hours of incubation with gastrin, cells were lysed with 1× luciferase lysis buffer (Toyo Ink, Inc, Tokyo, Japan). Luciferase activity was measured using PicaGene reagent kit (Toyo Ink) in Lumat LB9501 luminometer (Berthold, Wildbad, Germany). Enzyme activity was normalised for efficiency of transfection on the basis of SeaPansy luciferase activity and relative values were determined. The transfection experiments were carried out six times independently and the average of the values was calculated. In some examinations cells were preincubated with AG1478, PD98059, SB203580, GF109203X, L-740,093, Go6976, or rottlerin for one hour prior to gastrin stimulation. In other experiments cells were cotransfected with the plasmid encoding dominant negative IKKα, IKKβ, NIK, TRAF2, TRAF6, TAK1, PKC-θ, or PKC-δ.
Nuclear extract preparation and electrophoretic mobility shift assay (EMSA)
Cells were stimulated in the presence or absence of 10−8 M gastrin for 0.5 hours. Nuclear extracts from cells were prepared using a modification of Digman’s procedure.29 In some experiments cells were preincubated with AG1478, PD98059, SB203580, GF109203X, L-740,093, HBDDE, or rottlerin for one hour, or were transfected with the plasmid encoding the dominant negative IKKα, IKKβ, NIK, TRAF2, TRAF6, PKC-θ, or PKC-δ, 24 hours before gastrin stimulation. Protein concentrations were determined using BCA Protein Assay Reagent (Pierce, Rockford, Illinois, USA). EMSA for NFκB was performed by Gel Shift Assay Systems (Promega) according to the manufacture’s guidelines. A double stranded oligonucleotide probe for NFκB (5‘-AGTTGAGGGGACTTTCCCAGGC-3‘) was end labelled with [γ-32P] ATP. Each 10 μg of nuclear protein was incubated with a 32P labelled probe (5×104 cpm/reaction) and 0.5 μg/ml poly (dI-dC) poly (dI-dC) in 10 μl of binding buffer (4% glycerol, 1 mM MgCl2, 0.5 mM ethylenediamine tetraacetic acid (EDTA), 0.5 mM DTT, 50 mM NaCl, 10 mM Tris HCl, pH 7.5) for 20 minutes at room temperature. Samples were then loaded onto a 4% polyacrylamide gel (acryoamid/N, N‘-methylene bisacryoamide, 30:1) with 0.5% Tris borate EDTA buffer. After electrophoresis, gels were dried and exposed to Kodak XOMAT AR film (Eastman Kodak, Rochester, New York, USA).
Western blot analysis
Cells were grown to subconfluence in 60 mm dishes and deprived of serum for 24 hours. Thereafter they were stimulated with 10−8 M gastrin for 5, 15, 30, or 60 minutes and then lysed in lysis buffer containing 20 mM Tris (pH 8.0), 137 mM NaCl, 10% glycerol, 1% Nonidet P-40, 10 mM EDTA, 100 mM NaF, 1 mM PMSF, 0.25 TIU/ml aprotinin, and 10 mg/ml leupeptin. Aliquots containing 50 μg of total protein were size fractionated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (5–20% gradient gels), and proteins were transferred to polyvinylidine difluoride membranes (Immobilon; Millipore, Bedford, Massachusetts, USA). Membranes were blocked with Block Ace (Dainihon, Osaka, Japan) and incubated for 12 hours at 4°C with rabbit antihuman-phospho-PKC-δ (Thr505) polyclonal antibody (Cell Signaling Technology, Inc.) or rabbit antihuman-IκB-α antibody (Santa Cruz Biotechnology Inc.). After three washings with 0.1% Tween 20 in Tris buffered saline, membranes were incubated for one hour at room temperature with peroxidase conjugated goat antirabbit IgG (N Pharmaceuticals Inc., Aurora, Ohio, USA). After washing the membranes again, peroxidase was detected with an enhanced chemiluminescence system (ECL; Amersham). The protein concentrations of the homogenates were determined using BCA Protein Assay Reagent (Pierce).
Isolated parietal cells
Guinea pig isolated parietal cells were prepared and enriched essentially as described previously.11 Briefly, two male guinea pigs weighing approximately 300 g were anaesthetised with pentobarbital before sacrifice and their glandular stomachs were then excised. The fundic mucosa was scraped off and placed in 40 ml of buffer A (0.5 mM NaH2PO4, 1.0 mM Na2HPO4, 70 mM NaCl, 5 mM KCl, 11 mM glucose, 2 mM EDTA, 50 mM HEPES (pH 7.2), 20 mM NaHCO3, and 2% bovine serum albumin (BSA)) with 2.5 mg/ml pronase (Kaken Pharmaceutical Co. Ltd, Tokyo, Japan), which was subsequently incubated under gentle stirring for 15 minutes at 37°C. Mucosal fragments were then centrifuged for one minute at 250 g at room temperature. The supernatant was discarded and the remaining tissue pieces were resuspended in 40 ml of buffer B (1.0 mM CaCl2 and 1.5 mM MgCl2 instead of the EDTA in buffer A), which was further incubated under constant gassing with 95% O2/5% CO2. The incubation mixture was then filtered through a nylon cloth and centrifuged, followed by resuspension in RPMI-1640 medium containing 0.1% BSA and 10 mM HEPES (pH 7.2). Separation of the different cell populations was achieved by counterflow centrifugation in an elutriation system (SRR6Y; Hitachi Industrial, Tokyo, Japan). The cell suspension was loaded at a flow rate of 10 ml/min into the elutriation chamber, running at 1800 rpm. Seven different cell fractions (F1–F7) were separated at the following flow rates: F1, 10 ml/min; F2, 15 ml/min; F3, 20 ml/min; F4, 25 ml/min; F5, 30 ml/min; F6, 40 ml/min; and F7, 80 ml/min. Cells in F6 and F7 were collected, resuspended in serum free RPMI medium containing 5 mM N-acetylcysteine (Sigma). After two hours of pretreatment with GF109203X or rottlerin, cells were incubated with 10−8 M gastrin for 30minutes. The nuclear extracts were collected and subjected to EMSA as described above.
Statistics
Data are expressed as means (SEM). Statistical comparisons were made using the Student’s t test. A p level of <0.05 was considered to be statistically significant.
RESULTS
Activation of NFκB by gastrin and its inhibition by gastrin receptor antagonist and PKC inhibitor
The effect of gastrin on the binding activities of NFκB was examined by EMSA (fig 1A⇓). NFκB specific DNA-protein complexes were observed in nuclear proteins of MKGR26 cells that had been treated with gastrin. However, this effect was abolished by pretreatment with the gastrin receptor antagonist L-740,093. We also examined the effects of the PKC inhibitor GF109203X, the MEK inhibitor PD98059, the p38 MAPK inhibitor SB203580, and tyrphostin AG1478 on NFκB specific DNA-protein complex formation. Pretreatment with GF109203X abolished this gastrin induced complex formation whereas AG1478, PD98059, and SB203580 had no effect on NFκB binding activity (fig 1A⇓).

Effects of tyrphostin AG1478, the protein kinase C inhibitor GF109203X, the mitogen activated protein kinase kinase inhibitor PD98059, the p38 mitogen activated protein kinase inhibitor SB203580, and the gastrin receptor antagonist L-740,093 on gastrin induced activation of nuclear factor κB (NFκB), assessed by electrophoretic mobility shift assay (EMSA). (A) After incubation with the inhibitors for one hour, MKGR26 cells were treated with gastrin for 0.5 hours. Nuclear cell extracts of MKGR26 cells were assayed for NFκB DNA binding activity using the labelled oligonucleotide probes. (B) MKGR26 cells were transfected with pNFκB-LUC and pRL-SV40 as an internal standard. After preincubation with the inhibitors for one hour they were treated with gastrin for eight hours and the luciferase assay was performed. The results are expressed as fold induction compared with control cells treated with vehicle alone. Values are means (SEM) from six independent experiments (*p<0.05 v bar 1; †p<0.05 v bar 2).
We next performed luciferase assays with MKGR26 cells using the reporter gene pNFκB-LUC containing five tandem repeats of the NFκB binding sites. As shown in fig 1B⇑, GF109203X suppressed gastrin induced luciferase activity but no effect was detected for AG1478, PD98059, or SB203580. This is in general agreement with the EMSA results.
Involvement of PKC-δ in gastrin induced activation of NFκB
The implication of PKC in the activation of NFκB by gastrin prompted us to determine which PKC isozymes are responsible for inducing this phenomenon. We first examined the effect of PKC inhibitors on NFκB specific DNA-protein complex formation by gastrin. Rottlerin, a known inhibitor of PKC-δ and PKC-θ, partially inhibited DNA-protein complex formation (fig 2A⇓). In contrast, HBDDE, an inhibitor of PKC-α and PKC-γ, had no effect on complex formation (fig 2A⇓). We next determined the effect of rottlerin and that of Go6976, an inhibitor of PKC-α and PKC-β, on luciferase activities using pNFκB-LUC. Consistent with the EMSA results, rottlerin inhibited gastrin induced transcriptional activity of NFκB in MKGR26 cells. However, Go6976 had no effect on this process (fig 2B⇓). We also confirmed that gastrin activated NFκB in guinea pig isolated parietal cells and that GF109203X and rottlerin reduced gastrin induced NFκB binding activity (fig 2C⇓). As rottlerin inhibited NFκB activation by gastrin, the issue of whether the introduction of a dominant negative mutant of PKC-δ or of PKC-θ would inhibit gastrin induced NFκB luciferase activity was examined in MKGR26 cells. As a result, the dominant negative (dn) PKC-δ abrogated gastrin stimulated NFκB activation while dnPKC-θ had no effect (fig 3⇓).

Effects of protein kinase C inhibitors on gastrin induced activation of nuclear factor κB (NFκB). (A) After incubation with rottlerin or HBDDE for one hour, MKGR26 cells were treated with gastrin for 0.5 hours. Nuclear cell extracts of MKGR26 cells were assayed for NFκB DNA binding activity using the labelled oligonucleotide probe. (B) MKGR26 cells were transfected with pNFκB-LUC and pRL-SV40 as an internal standard. After preincubation with rottlerin or Go6976 for one hour, they were treated with gastrin for eight hours and the luciferase assay was performed. The results are expressed as fold induction compared with control cells treated with vehicle alone. Values are means (SEM) from six independent experiments (*p<0.05 v bar 1; †p<0.05 v bar 2). (C) Guinea pig parietal cells were isolated and incubated with or without GF109203X or rottlerin for two hours. Then, cells were treated with gastrin for 0.5 hours and the nuclear cell extracts were assayed for NFκB DNA binding activity using the labelled oligonucleotide probe.

Effects of protein kinase C (PKC) isozyme mutants on gastrin induced nuclear factor κB (NFκB) activation. MKGR26 cells were cotransfected with 0.5 μg of pNFκB-LUC and a construct encoding the dominant negative (dn) mutant of PKC-δ or PKC-θ. After 24 hours, cells were treated with gastrin for eight hours and the luciferase assay was performed. The results are expressed as fold induction compared with control cells treated with vehicle alone. Values are means (SEM) from six independent experiments (*p<0.05 v bar 1; †p<0.05 v bar 2).
Gastrin phosphorylates PKC-δ in MKGR26 cells
Because PKC-δ appears to mediate gastrin induced activation of NFκB, we next determined whether gastrin induces phosphorylation of PKC-δ by western blot analysis using phospho-PKC-δ antibody. As shown in fig 4⇓, gastrin induced phosphorylation of PKC-δ in MKGR26 cells in a time dependent manner, while it was not observed in MKN-neo cells. The phosphorylated form of PKC-δ was detected within five minutes after the start of gastrin stimulation and the level of phosphorylation peaked at 15 minutes and then remained for over 60 minutes.
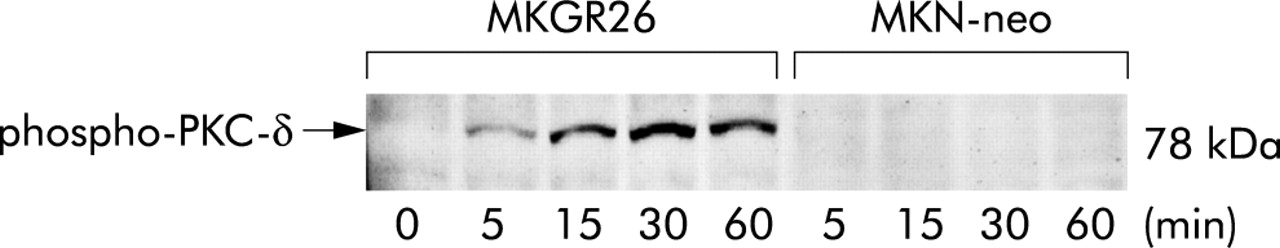
Gastrin induced phosphorylation of protein kinase C (PKC)-δ. MKGR26 cells and MKN-neo cells were treated with gastrin for 0, 5, 15, 30, or 60 minutes and cell lysates were then obtained. Proteins were size fractionated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis and transferred to membranes. Membranes were reacted with rabbit antihuman-phospho-PKC-δ (Thr505) polyclonal antibody, incubated with peroxidase conjugated goat antirabbit IgG, and peroxidase was detected with an enhanced chemiluminescence system.
Gastrin induces degradation of IκB
It is well known that both IL-1 and TNF-α induce activation of NFκB via the phosphorylation dependent degradation of IκBs.16 Immunoblot analysis was performed using anti-IκB-α antibody to determine whether gastrin induces degradation of IκB-α. A decrease in IκB-α protein was observed within five minutes after initiation of gastrin stimulation in MKGR26 cells while we could not find degradation in MKN-neo cells (fig 5⇓).

Gastrin induced degradation of inhibitor κB (IκB)-α. MKGR26 cells and MKN-neo cells were treated with gastrin for 0, 5, 15, or 30 minutes and cell lysates were obtained. Proteins were size fractionated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis, and then transferred to polyvinylidine difluoride membranes. Membranes were reacted with rabbit antihuman-IκB-α antibody, incubated with peroxidase conjugated goat antirabbit IgG, and peroxidase was detected with an enhanced chemiluminescence system. Quantitated signals were determined by densitometry.
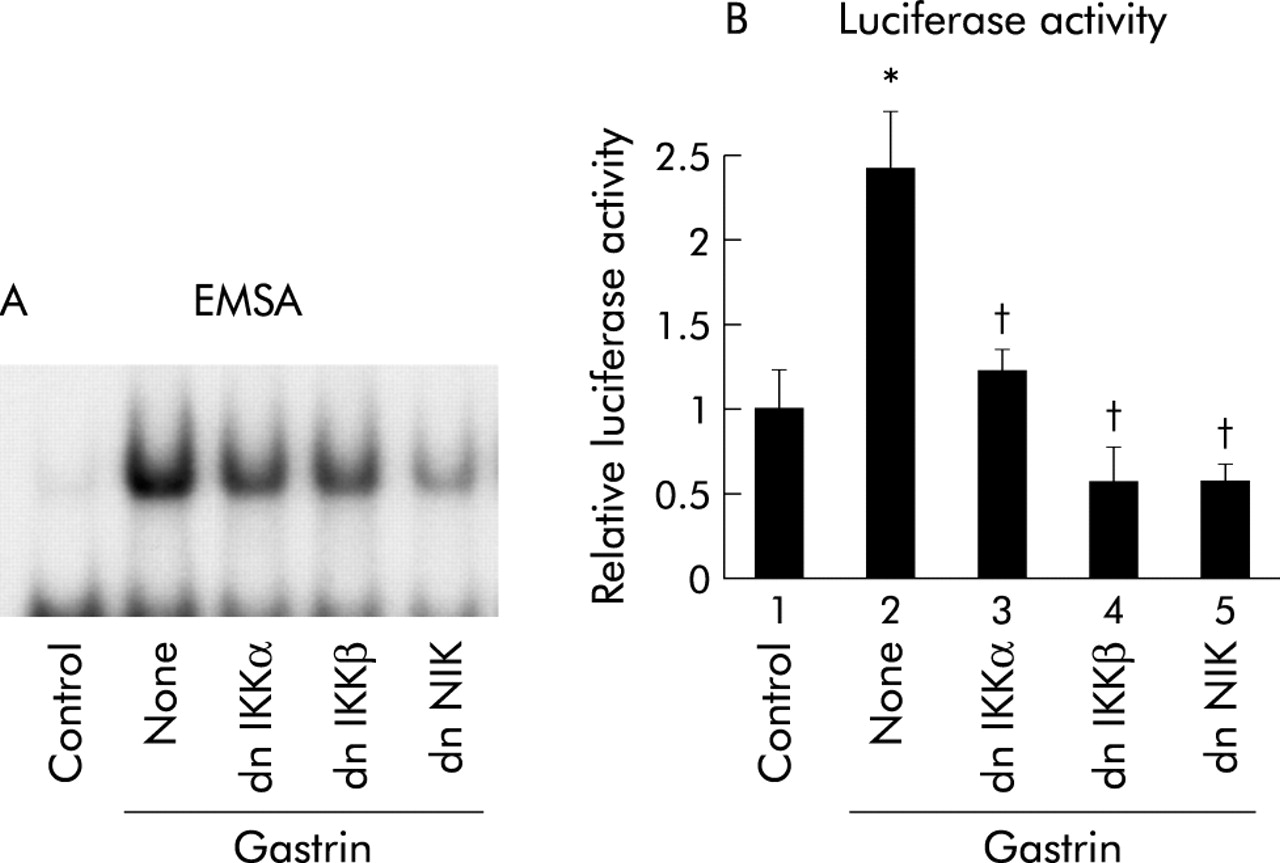
Kinase defective IKKs and NIK inhibit NFκB activation by gastrin
As phosphorylation of IκB by IKK is required for its degradation,12,13 we determined the involvement of IKKs in gastrin induced NFκB activation. EMSA was performed to evaluate the involvement of IKKs in NFκB specific DNA-protein complex formation by gastrin. MKGR26 cells were transfected with 0.5 μg of dnIKKα or dnIKKβ prior to gastrin stimulation. Expression of dnIKKα or dnIKKβ inhibited gastrin induced NFκB binding activity, indicating that IKKs are required for the response (fig 6A⇓). Because the IKK complex is activated by NIK,21 involvement of NIK in NFκB activation by gastrin was examined. Coexpression of dnNIK inhibited gastrin induced NFκB activation (fig 6A⇓).

Effects of inhibition of inhibitor κB kinase (IKK) or NFκB inducing kinase (NIK) on gastrin induced nuclear factor κB (NFκB) activation. (A) MKGR26 cells were transfected with 1 μg of pRK5 or a construct encoding the dominant negative (dn) mutant of IKKα, IKKβ, or NIK. After 24 hours, MKGR26 cells were treated with gastrin for 0.5 hours. Nuclear cell extracts of MKGR26 cells were assayed for NFκB DNA binding activity using the labelled oligonucleotide probe. (B) MKGR26 cells were transfected with pNFκB-LUC and pRK5 or a construct encoding the dominant negative (dn) mutant of IKKα, IKKβ, or NIK. pRL-SV40 was cotransfected as an internal standard. After 24 hours they were treated with gastrin for eight hours and the luciferase assay was performed. Results are expressed as fold induction compared with control cells treated with vehicle alone. Values are means (SEM) from six independent (*p<0.05 v bar 1; †p<0.05 v bar 2).
We also performed luciferase assays to evaluate the involvement of IKKs and NIK in activation of NFκB by gastrin. MKGR26 cells were cotransfected with pNFκB-LUC in combination with dnIKKα, dnIKKβ, or dnNIK. As shown in fig 6B⇑, introduction of dnIKKα, dnIKKβ, or dnNIK reduced the gastrin induced binding activity of NFκB.
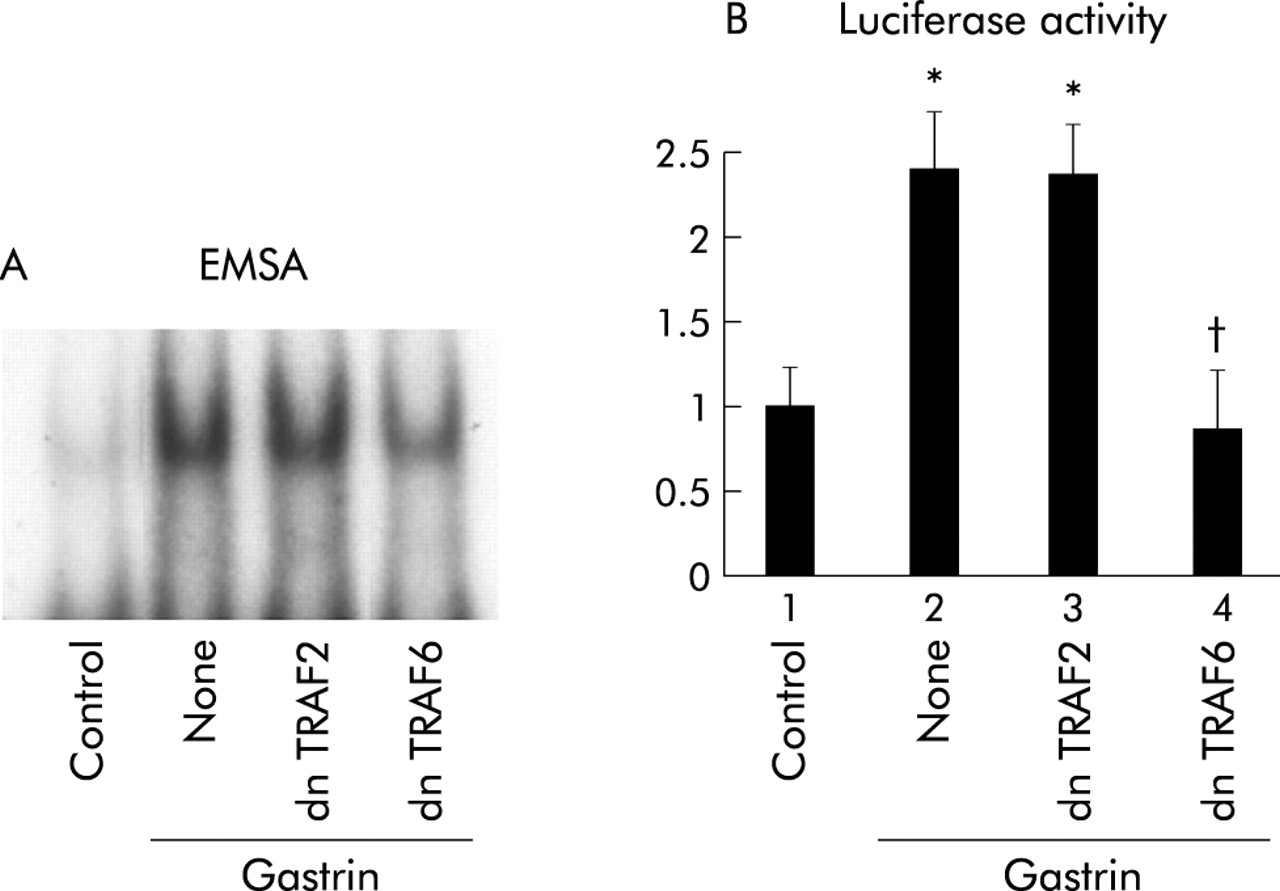
Dominant negative TRAF6 but not TRAF2 inhibits gastrin induced NFκB activation
TRAF6 and TRAF2 are known to be involved in the activation of NIK in IL-1 and TNF receptor mediated signals, respectively.19,20 As a result, we determined whether TRAF2 and TRAF6 are involved in the gastrin receptor mediated signalling pathway. Coexpression of dnTRAF6 inhibited NFκB specific DNA-protein complex formation while dnTRAF2 had no effect (fig 7A⇓). Consistent with the EMSA results, introduction of dnTRAF6 caused a reduction in gastrin induced NFκB activity while dnTRAF2 had no effect (fig 7B⇓).
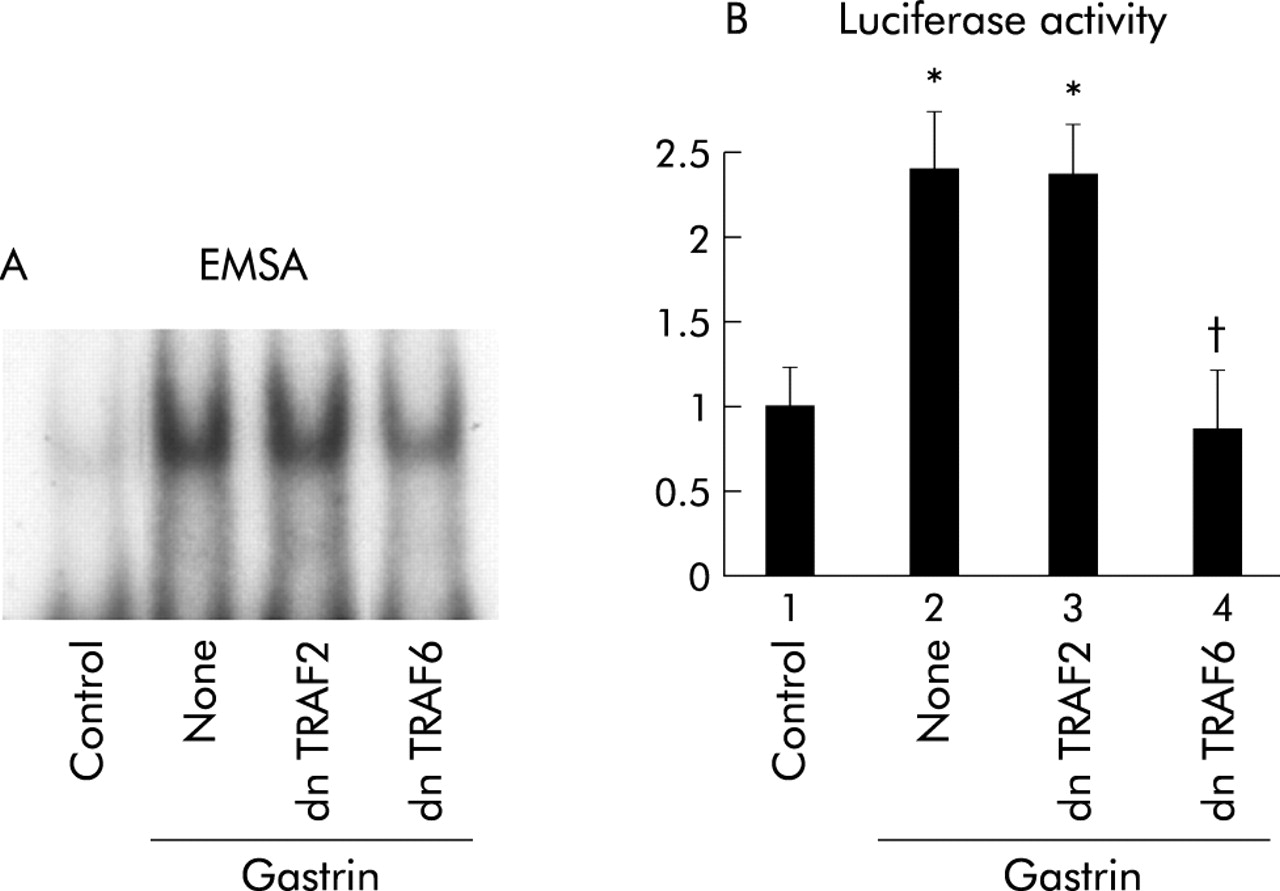
Effects of inhibition of tumour necrosis factor receptor associated factor (TRAF) 2 or TRAF6 on gastrin induced nuclear factor κB (NFκB) activation. (A) MKGR26 cells were transfected with 1 μg of pRK5 or a construct encoding the dominant negative (dn) mutant of TRAF2 or TRAF6. After 24 hours cells were treated with gastrin for 0.5 hours. Nuclear cell extracts of MKGR26 cells were assayed for NFκB DNA binding activity using the labelled oligonucleotide probe. (B) MKGR26 cells were transfected with pNFκB-LUC and pRK5 or a construct encoding the dominant negative (dn) mutant of TRAF2 or TRAF6. pRL-SV40 was cotransfected as an internal standard. After 24 hours they were treated with gastrin for eight hours and the luciferase assay was performed. Results are expressed as fold induction compared with control cells treated with vehicle alone. Values are means (SEM) from six independent (*p<0.05 v bar 1; †p<0.05 v bar 2).
Because TAK1 has been reported to associate with TRAF6 and to activate NIK in the IL-1 signalling pathway,21,22 we next examined the involvement of TAK1 in this pathway. As shown in fig 8⇓, dnTAK1 inhibited gastrin induced NFκB luciferase activity.

Effect of the dominant negative transforming growth factor β activated kinase 1 (dnTAK1) on gastrin induced nuclear factor κB (NFκB) activation. MKGR26 cells were transfected with pNFκB-LUC and control vector or a construct encoding the dominant negative mutant of TAK1. pRL-SV40 was cotransfected as an internal standard. After 24 hours they were treated with gastrin for eight hours and the luciferase assay was performed. The results are expressed as fold induction compared with control cells treated with vehicle alone. Values are means (SEM) from six independent (*p<0.05 v bar 1; †p<0.05 v bar 2).
Activation of NFκB by PKC-δ and its inhibition by dominant negative TRAF6, NIK, or IKKs
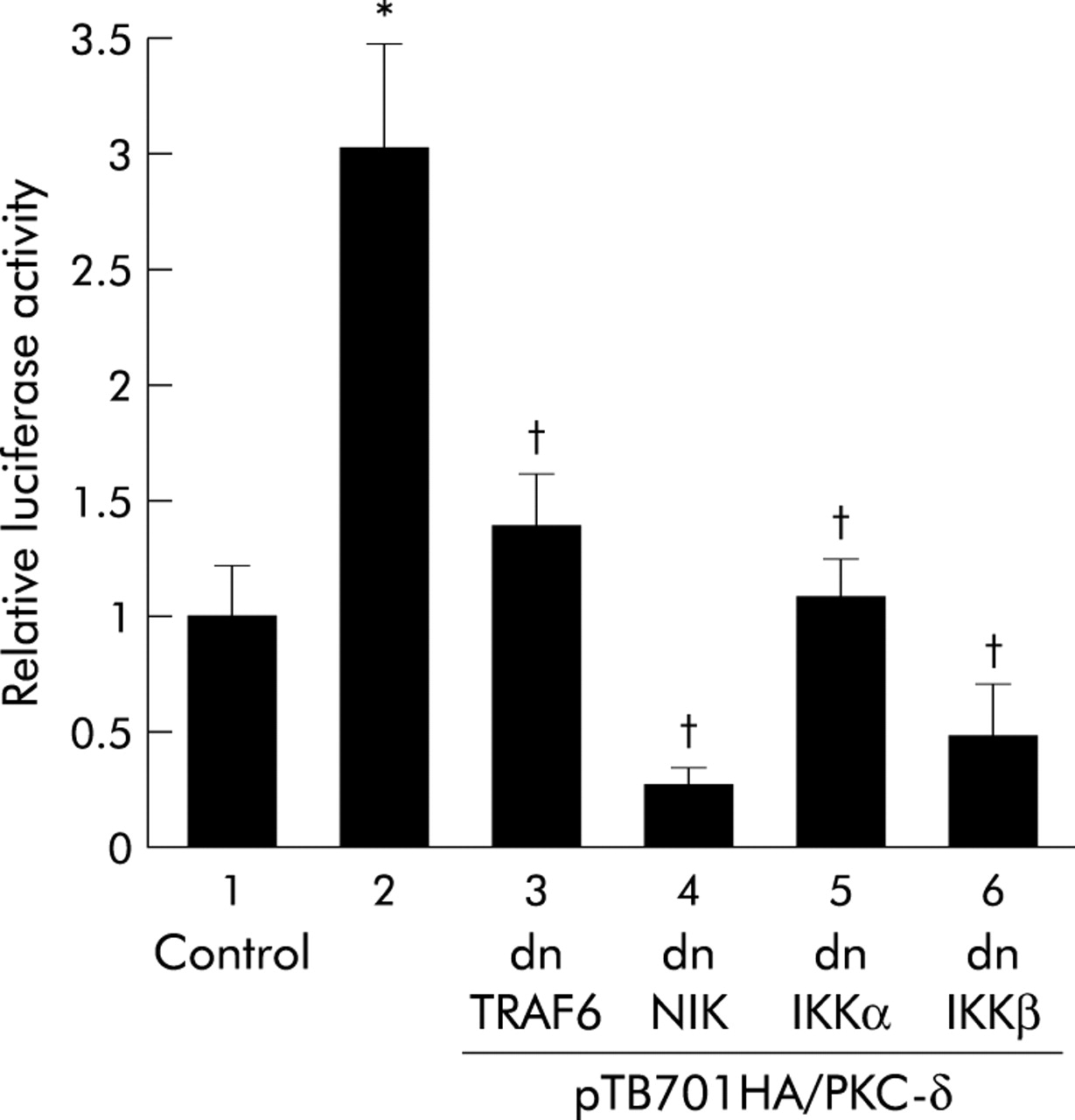
The issue of whether forced expression of PKC-δ alone activates NFκB in MKGR26 cells was examined. MKGR26 cells were cotransfected with pNFκB-LUC and pTB701HA/PKC-δ, a construct encoding wild-type PKC-δ. As shown in fig 9⇓, forced expression of PKC-δ induced NFκB activation. However, this effect was inhibited by cotransfection of the dominant negative mutant of TRAF6, NIK, or IKKs (fig 9⇓).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effects of inhibition of inhibitor κB kinase (IKK), NFκB inducing kinase (NIK), or tumour necrosis factor receptor associated factor (TRAF) 6 on nuclear factor κB (NFκB) activation induced by forced expression of protein kinase C (PKC)-δ. MKGR26 cells were cotransfected with 0.5 μg of pNFκB-LUC, pTB701HA/PKC-δ, a construct encoding wild-type PKC-δ, and the dominant negative (dn) mutant of IKKα, IKKβ, NIK, or TRAF6. After 24 hours, the luciferase assay was performed. Values are means (SEM) from six independent (*p<0.05 v bar 1; †p<0.05 v bar 2).
DISCUSSION
The present study clearly demonstrates that gastrin is capable of activating NFκB, one of the most important transcription factors. Activation of NFκB in gastrin receptor transfected cells was determined by both NFκB specific DNA-protein complex formation and luciferase activity using a reporter gene containing five tandem repeats of the NFκB binding sites. The data obtained from the two different methods were in general agreement and gastrin induced NFκB activation was abolished by pretreatment with L-740,093, indicating that this is a gastrin receptor mediated reaction. Our study also showed that gastrin is capable of inducing formation of NFκB specific DNA-protein complex in isolated parietal cells, suggesting that this effect of gastrin is of physiological significance.
The gastrin receptor is a member of the family of GPCRs that are characterised by a seven transmembrane domain structure.6,7 Leucocytes, mesenchymal cells, and epithelial cells express various cell type specific GPCRs30–,33; their involvement in physiological functions has been verified. Recent studies have shown that NFκB is involved in GPCR mediated biological functions in leucocytes, smooth muscle cells, and endothelial cells. For example, cytokines and chemoattractants that bind to GPCRs are known to induce production of proinflammatory cytokines and chemokines via activation of NFκB in leucocytes.30,31 Thrombin activates NFκB which is associated with smooth muscle cell proliferation, enhanced ICAM-1 expression, and neutrophil adhesion.31,33 The gastrin/NFκB pathway may also play physiological roles in the gastric mucosa. One possible role is modulating chemokine production in gastric epithelial cells. Our previous in vitro study showed that gastrin induces IL-8 production synergistically with Helicobacter pylori.2 This may account for increased IL-8 production in gastric mucosa of H pylori infected subjects with hypergastrinaemia34–,36 and enhancement of gastric inflammation by gastrin in a Helicobacter infected animal model.37 Another possible role is regulating gastric mucosal growth and differentiation which is a well known function of gastrin.1 Interestingly, mice lacking the COOH terminal ankyrin domain of NFκB2 showed marked gastric hyperplasia, suggesting the involvement of the NFκB family in gastric epithelial cell growth and/or apoptosis.38
Because gastrin receptor mediated signals are associated with several protein kinases,7–,11 we determined the specific kinases that are involved in gastrin activation of NFκB. Of the kinase inhibitors tested, the general PKC inhibitor GF109203X abolished gastrin induced NFκB activation. The PKC superfamily consists of three subfamilies—that is, conventional PKC (PKC-α, -β, and -γ), novel PKC (PKC-δ, -ε, -μ, -θ and -η), and atypical PKC (PKC-ζ and -λ/ι).39 Recent investigations have proposed that different PKC isoforms are involved in NFκB activation depending on the cell type. For example, PKC-θ is required for T cell receptor induced NFκB activation40 and PKC-δ is associated with thrombin induced NFκB activation in endothelial cells.33 In the present study, gastrin induced phosphorylation of PKC-δ and its inhibition suppressed gastrin induced NFκB activation partially. Furthermore, forced expression of PKC-δ alone is capable of inducing NFκB activation in MKGR26 cells. This suggests that PKC-δ plays a role in the gastrin/NFκB pathway in gastric epithelial cells, at least in part.
IL-1 and TNF-α activate NFκB in most cell types through the NIK/IKK/IκB signalling pathway.20–,22 Our study showed that gastrin induced degradation of IκB and that the introduction of kinase defective NIK or IKKs inhibited NFκB activation by gastrin. These results indicate that gastrin also uses the NIK/IKK/IκB pathway to activate NFκB. Signalling pathways upstream of NIK differ depending on the receptors involved. Reportedly, IL-1 receptor forms a complex with adaptor proteins Myd88 and IRAK, which recruit TRAF6,41,42 while TNF-α receptor binds to adapter proteins TRADD and RIP, and the complex recruits TRAF2.43 Although both TRAF2 and TRAF6 can activate NIK,19,20 the present study suggests that TRAF6, but not TRAF2, is required for gastrin induced NFκB activation. In addition, TAK1, which interacts with TRAF6 in IL-1 signalling,21 is also required for gastrin induced NFκB activation, suggesting that signalling of gastrin is similar to that of IL-1.
As shown in figs 6 and 7⇑⇑, the inhibitory effects of the dominant negative TRAF6, NIK, and IKKs on gastrin induced NFκB activity appeared to be less by EMSA than by luciferase assays. This may be explained as follows. Because each dominant negative plasmid was introduced into the cells along with the reporter gene pNFκB-LUC, it is reasonable to suggest that the transfection efficiency would not affect its inhibitory effects assessed by luciferase assays. In contrast, it is likely that the inhibitory effects of the dominant negative plasmids on NFκB DNA complex formation evaluated by EMSA would entirely depend on the transfection efficiency. The low transfection efficiency may have obscured the full inhibitory effects in EMSA, as shown in figs 6 and 7⇑⇑. In fig 6⇑, inhibition of IKKβ appears to be less effective than that of NIK when tested by EMSA although these inhibitory effects are similar when tested by luciferase assay. This may also be explained by the difference in transfection efficiency between the two plasmids.
Our study also showed that forced expression of PKC-δ alone induced activation of NFκB while inactivation of TRAF6, NIK, or IKKs inhibited this effect. Thus PKC-δ may be directly or indirectly linked to NIK/ IKK/IκB signalling. Reportedly, p38 MAPK plays a role downstream of PKC-δ in thrombin induced activation of NFκB in endothelial cells.33 However, our data do not suggest that the same mechanism is operative in the gastrin/NFκB pathway. Although one report indicated that PKC-ζ is capable of interacting with IRAK directly,44 we could not find a direct association of PKC-δ with IRAK or TRAF6 (unpublished observation). Thus the pathway that links PKC-δ to NIK/ IKK/IκB signalling is still unknown.
In conclusion, our results indicate that gastrin is capable of activating NFκB via a PKC dependent pathway, which is directly or indirectly linked to the NIK/IKK/IκB signalling pathway. Further studies are needed to clarify the pathophysiological significance of this signalling in hypergastrinaemia.
Acknowledgments
The authors thank Dr M Takeuchi (Yamanouchi Pharmaceutical Co. Ltd, Tsukuba, Japan) for skillful discussions.