Article Text

Abstract
Background Anti-tumour necrosis factor α (TNFα) therapy effectively induces and maintains remission in Crohn's disease (CD). Up to 40% of patients, however, fail to respond to anti-TNFα.
Objective To identify the mechanisms underlying the persistence of mucosal lesions in patients who fail to respond to anti-TNFα therapy.
Design An observational study based on whole-genome transcriptional analysis was carried out using intestinal biopsy specimens from patients with CD receiving (n=12) or not (n=10) anti-TNFα therapy. The transcriptional signature of responders was compared with that of non-responders after anti-TNFα therapy. Controls with non-inflammatory bowel disease (non-IBD) (n=17) were used for comparisons. Genes of interest were validated by real-time RT-PCR in an independent cohort of patients with CD receiving (n=17) or not (n=16) anti-TNFα and non-IBD controls (n=7).
Results We confirmed that response to anti-TNFα is accompanied by significant regulation of a large number of genes, including IL1B, S100A8, CXCL1, which correlated with endoscopic activity. Remarkably, patients who failed to respond to anti-TNFα showed a mixed signature, maintaining increased expression of IL1B, IL17A and S100A8, while showing significant modulation of other genes commonly upregulated in active CD, including IL6 and IL23p19.
Conclusions Our results show that anti-TNFα therapy significantly downregulates a subset of inflammatory genes even in patients who fail to achieve endoscopic remission, suggesting that these genes may not be dominant in driving inflammation in non-responders. On the other hand, we identified IL1B and IL17A as genes that remained altered in non-responders, pointing to potentially more relevant targets for modulating mucosal damage in refractory patients.
- CROHN'S DISEASE
- TNF-ALPHA
- INFLAMMATORY MEDIATORS
- IBD BASIC RESEARCH
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
-
Anti-tumour necrosis factor α (anti-TNFα) antibodies are effective in managing Crohn's disease and for many patients, provide a therapeutic alternative.
-
40% of patients will not respond to this treatment or will have a loss of response that is not related to immunogenicity.
-
The mechanisms sustaining active inflammation in inflammatory bowel disease in the presence of effective TNFα blockade have not been established.
What are the new findings?
-
Anti-TNFα therapy regulates a subset of inflammation-dependent genes, including IL6 and IL23p19, regardless of response to treatment.
-
Modulation of TNFα-dependent genes in non-responsive patients suggests that lack of a response to anti-TNFα does not result from the absence of biological activity of the administered antibody.
-
Patients who are refractory to anti-TNFα therapy maintain deregulated transcription of a set of genes including IL1B, S100A8, S100A9, CXCL2, CXCL6 and S100A12.
-
Pathway analysis of TNFα-resistant genes identified IL17A as a potential mediator driving anti-TNFα refractory inflammation.
How might it impact on clinical practice in the foreseeable future?
-
Genes that remained altered in patients not responding to TNF blockade (ie, IL1B, IL17A) may be effective targets for inducing remission in this group of patients with refractory disease.
-
In contrast, the targeting of mediators, such as IL6, which are downregulated by anti-TNFα treatment even in non-responders, may not provide additional benefits to patients with refractory disease.
Introduction
Crohn's disease (CD) is a chronic inflammatory disease of the intestinal tract with a relapsing course. The main goals of CD management are to achieve and maintain clinical and endoscopic remission and to avoid the development of complications such as stenosis and fistulae. Treatment targets the immune and inflammatory response and involves conventional drugs such as corticosteroids and azathioprine and the more recently introduced anti-tumour necrosis factor α (anti-TNFα) antibodies.1–3
For more than a decade, studies have shown that antibodies against TNFα can induce and maintain remission in patients with CD, with about 60% of patients responding.4 The percentage of patients who respond to a second anti-TNFα agent, however, is lower than with the first agent.5 ,6 Lack of response may stem from insufficient dose (low serum drug levels), development of antibodies against the drug (immunogenicity), or the presence of predominantly fibrotic strictures.7 An alternative explanation, however, is that in some patients, mechanisms that are independent of TNFα signalling may be driving inflammation. Such mechanisms may affect each patient differently and may even change as the disease progresses.8
To better understand the mechanisms underlying sustained inflammation in patients treated with anti-TNFα antibodies, we compared the transcriptional signature in intestinal biopsy specimens from patients with CD receiving anti-TNFα therapy based on their endoscopic response to treatment. Using this approach, we identified potential mediators of persistent inflammation in non-responders (patients who did not achieve endoscopic remission after at least 12 weeks of anti-TNFα treatment). In addition, our study shows that even in non-responders, anti-TNFα antibodies do modulate a number of TNFα-dependent genes while maintaining inflammation. These observations strongly support the presence, in this group of patients, of redundant mechanisms contributing to disease, and also point towards potentially more selective targets.
Materials and methods
More detailed information is described in the online supplementary methods section.
Patient population
Patients with an established diagnosis of CD of more than 4 months’ duration and controls with non-inflammatory bowel disease (non-IBD) were included in the study from November 2010 to November 2013 after obtaining written informed consent. Non-IBD controls comprised subjects aged between 18 and 70 years undergoing colonoscopy for mild gastrointestinal symptoms or a screening colonoscopy for colorectal cancer and who had no lesions. All individuals were recruited at the Department of Gastroenterology, Hospital Clinic of Barcelona.
Clinical and demographic characteristics of patients with CD are represented in table 1. Endoscopic active disease was defined as a CD Endoscopic Index of Severity (CDEIS)9 of ≥5 and the presence of large ulcers (>0.5 cm diameter) in at least one of the explored segments. Clinical activity was defined as a CD Activity Index10 >150. C-reactive protein was determined in all patients. Patients were assessed according to the Montreal classification11 based on age at diagnosis (A), location (L) and behaviour (B) of the disease. Histology of all segments was reviewed by an expert pathologist (Hospital Clinic of Barcelona, Pathology Department). Patients with concomitant infections (Clostridium difficile, cytomegalovirus or Epstein–Barr virus) were excluded from the study. To exclude a pharmacokinetic basis for the non-response to anti-TNFα antibodies and to ensure adequate drug coverage, cases with low trough levels of drug (infliximab ≤2 μg/mL; adalimumab ≤4 μg/mL, based on analysis of our own large cohort) or any levels of anti-drug antibodies were excluded. Measurements of drug levels and antibodies were performed using the Promonitor assay (Progenika Spain), according to the manufacturer's instructions.
Clinical and demographic characteristics of patients included in cohorts 1 and 2
In this observational exploratory study a formal sample size calculation was not performed. Patients with CD were grouped into two independent cohorts. Cohort 1 (22 patients) was used for microarray analysis and cohort 2 (33 patients) for validation using real-time RT-PCR. In addition, 24 non-IBD controls (17 for microarray and 7 for real-time RT-PCR) who presented no lesions at colonoscopy were studied. Patients belonged to one of the following groups: endoscopically active CD with and without anti-TNFα therapy, endoscopically active patients with CD receiving anti-TNFα and non-IBD controls (table 1). Endoscopic response to anti-TNFα was assessed at 12 weeks after starting drug administration and endoscopic remission was defined as a CDEIS <5 and an absence of ulcers. A subset of patients with CD from cohort 1 (n=7), were followed up for 12 weeks after starting anti-TNFα therapy.
Patients were included in the study irrespective of the anti-TNFα antibody used. Adalimumab was given subcutaneously 160/80 mg at weeks 0/2 for induction and 40 mg every other week for maintenance. Infliximab was given intravenously (5 mg/kg) at weeks 0/2/6 for induction and every 8 weeks for maintenance.
Of those patients who did not respond to anti-TNFα therapy, but who were followed up for an additional 14 weeks, two had to undergo surgery, four were switched to a different class of drug and either achieved remission or milder disease and three were receiving higher doses of anti-TNFα but were still presenting mucosal lesions. Four patients are still receiving anti-TNFα at the standard doses and had mild disease at the time of follow-up. For two patients we have no follow-up information.
Tissue samples
Biopsy specimens (2–3) were taken from the colon of patients with CD or non-IBD controls during endoscopic evaluation. In patients with CD, samples were taken from the affected areas: in patients with active disease, samples were obtained from segments with endoscopic lesions and in patients with inactive disease from segments with previously documented involvement. The degree of inflammation assessed by endoscopic examination was confirmed by histological evaluation. No discrepancy between macroscopic and microscopic assessment was reported for any of the samples included in the study (data not shown).
Biopsy specimens were placed in RNA later-RNA Stabilisation Reagent (Qiagen, Spain) and stored at −80°C.
RNA isolation
Total RNA from biopsy specimens was extracted using an Rneasy Kit (Qiagen, Spain). Purity and integrity of the total RNA were assessed with the 2100 Bioanalyzer (Agilent, Germany) and then quantified by NanoDrop spectrophotometer (NanoDrop Technologies, USA); only samples with an RNA integrity number >7.0 were used.
Microarrays
The cRNA derived from biopsy specimens (150 ng) from cohort 1 (table 1) was hybridised to high-density oligonucleotide Affymetrix GeneChip Human Genome U133 Plus 2.0 Arrays (Affymetrix, USA) and raw data were analysed using Bioconductor tools in R (V.2.15.0) employing linear models for microarray data (LIMMA) for differential expression analysis. Pathway analysis of genes showing significant regulation was performed using Ingenuity Pathways Analysis (IPA; Ingenuity Systems).
Microarray raw data (.cel files) and processed data have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO series accession number GSE52746 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE52746).
Quantitative real-time RT-PCR
To validate the microarray data, we performed real-time RT-PCR analysis for three selected target genes (IL6, IL1B and S100A8) and one endogenous control (ACTB) using an independent group of 33 patients and seven non-IBD controls (table 1, cohort 2). Four additional target genes, IL23p19, IL12p35, IL17A and IL17F were assessed by real-time RT-PCR.
Microarray and real-time RT-PCR data correlation was assessed in a subset of 14 tissue samples (four active CD without anti-TNFα, four active CD with anti-TNFα, three inactive CD with anti-TNFα and three non-IBD controls) used for microarray analysis (table 1, cohort 1). High correlation between microarray and real-time RT-PCR data, r > 0.9 and p value <0.001, was seen for all three genes (data not shown).
Statistical analysis
Continuous variables were described as median and numerical range (min, max) and categorical variables as absolute frequencies. A non-parametric Wilcoxon test was performed to analyse the statistical significance of differences between groups for clinical variables and real-time RT-PCR values. The Benjamini–Hochberg procedure was used for multiple testing correction.12 A p value of <0.05 was considered statistically significant. The Pearson's product moment correlation to test for any associations between paired data was applied. All analyses were performed using R statistical environment (V.2.15.0).
Ethical considerations
This study was performed in accordance with the Declaration of Helsinki and was approved by the institutional ethics committee of the Hospital Clinic Barcelona (25 November 2010, Spain).
Results
Whole transcriptional analysis identifies two different subsets of patients with CD based on anti-TNFα response
Microarray data from all patients included in cohort 1 (table 1) were analysed in order to understand the mechanisms underlying their lack of response to anti-TNFα therapy. First, we compared the transcriptional profiles of patients with CD who did not achieve endoscopic healing despite treatment with anti-TNFα for 12 weeks (non-responders) with those who showed mucosal healing (responders) after anti-TNFα therapy.
Principal component analysis from whole microarray transcriptomic data (14 169 genes) showed a clear segregation of samples from patients with active and inactive CD (figure 1). Samples from patients who achieved endoscopic remission with anti-TNFα treatment (in blue) were clustered together with samples from non-IBD controls (in black), whereas samples from patients with active CD with (in green) or without anti-TNFα therapy (in red) remained separated. Nonetheless, this approach could not differentiate between patients with CD and inflammatory lesions based on whether or not they were receiving anti-TNFα therapy.

Principal component analysis of microarray-based genome-wide gene expression profiles derived from intestinal biopsy specimens. Mucosal biopsy specimens were obtained from controls with non-inflammatory bowel disease (non-IBD; black, n=17), patients with inactive Crohn's disease (CD) treated with anti-tumour necrosis factor α (anti-TNFα responders; blue, n=7), patients with active CD treated with anti-TNFα (non-responders, green, n=5) and patients with active CD not treated with anti-TNFα (red, n=10). A two-principal component plot is shown with the first component along the y axis and the second component along the x axis. The percentage of variability explained by each component is shown on the axis.
Response to anti-TNFα antibodies is associated with significant regulation of gene expression in the colonic mucosa of patients with CD
For a subset of patients with CD from cohort 1 (n=7), paired biopsy specimens, both before starting anti-TNFα therapy and at week 12 of therapy, were available. In these patients, anti-TNFα antibody administration induced clinical and endoscopic remission (CD Activity Index <150, CDEIS <5 and no ulcerations) in six out of the seven patients. In addition, a significant decrease in biomarkers of disease activity, including biological markers and the Magnetic Resonance Index of Activity (MaRIA), was seen at week 12 of anti-TNFα treatment in responders (table 2).
Clinical characteristics of anti-TNFα responders (cohort 1) whose tissue samples were analysed both before and after treatment (n=6)
Whole genome transcriptional analysis of colonic biopsy specimens at baseline (week 0) and after 12 weeks of anti-TNFα therapy identified 941 genes significantly regulated (688 downregulated) at week 12 compared with baseline in responders. Among these, 60 genes showed a fold-change >4 (|log2 ratio| ≥2). Figure 2 shows a heatmap representation of log2 ratios between week 0 and week 12 for all 60 genes in each individual patient (n=7). The one patient who showed no improvement either clinically or endoscopically in response to anti-TNFα (patient 7, in red, far right) can be recognised by the corresponding expression pattern. In contrast, patients who improved with treatment showed a marked regulation (downregulation at week 12 shown in blue, upregulation in red) of most of the selected 60 responsive genes. Transcription of those genes that were most sensitive to disease improvement showed a significant correlation with parameters of endoscopic and radiological disease activity. In contrast, correlation with other clinical parameters was not significant for most genes (see online supplementary table S1).
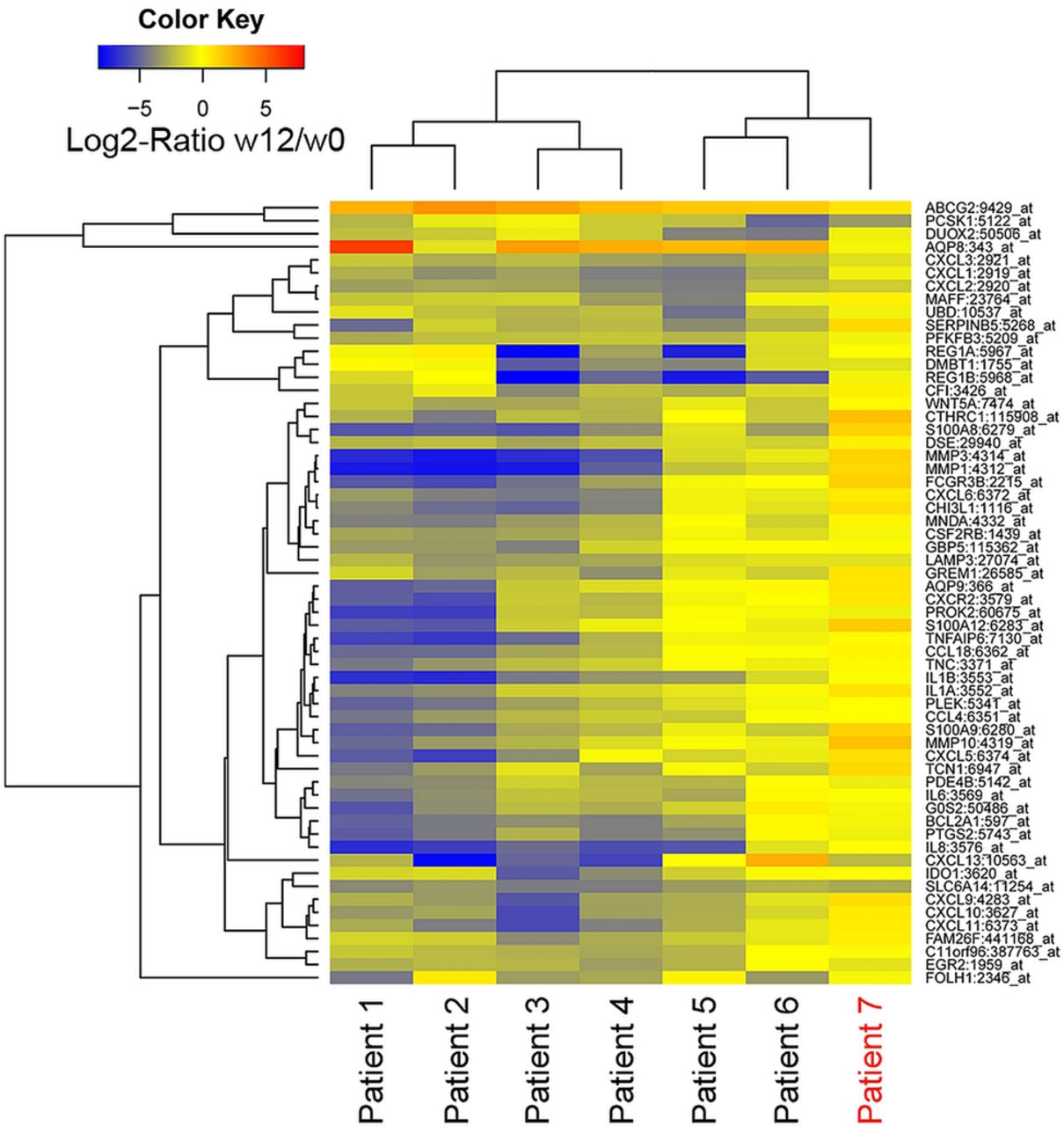
Heatmap representation of significant microarray gene expression fold-change after 12 weeks of anti-tumour necrosis factor α (anti-TNFα) therapy. Heatmap representation of microarray log2 fold-change of 60 target genes. Each row shows one individual gene and each column a patient with Crohn's disease. Changes in expression between weeks 12 and 0 of treatment are shown on a colour scale. Downregulated genes are shown in blue, unchanged genes in yellow and upregulated genes in red. An unsupervised hierarchical cluster method, using a Pearson distance and average linkage method, was applied for patients and genes classification. One patient who did not respond to anti-TNFα is labelled in red (patient 7).
Thus far our data confirm that induction of endoscopic remission by anti-TNFα administration is associated with significant modulation in the colonic mucosa of a large number of genes.
Analysis of microarray data reveals a unique transcriptional pattern in anti-TNF non-responders
While whole-genome principal component analysis could not identify patients with CD based on their anti-TNFα status (figure 1), statistical analysis of microarray data revealed a unique gene expression profile in anti-TNFα-treated patients, independent of their inflammatory state.
Analysis of microarrays (analysis of variance, unpaired, figure 3) identified 1310 genes whose expression was significantly regulated in the group of patients with active CD not receiving anti-TNFα therapy compared with non-IBD controls. Of those, 435 genes returned to control levels in responders, but remained deregulated in non-responders (figure 3A, genes denoted with one asterisk). Figure 3B shows the heatmap representation of a gene subset that fulfilled the additional criteria: (1) they were equally expressed in anti-TNFα non-responders and patients with active CD without anti-TNFα treatment and were also equally expressed in anti-TNFα responders and controls; and (2) their expression in anti-TNFα responders and non-responders differed significantly (see online supplementary table S2). This set of genes included REG1A, IL8, S100A8, S100A9, IL1B, MMP1 and MMP3 and represented those inflammation-dependent genes that were downregulated as mucosal healing was achieved. They were not, however, regulated in all patients receiving anti-TNFα; rather, only in those subjects who attained endoscopic remission.

Differentially expressed genes in the intestinal colonic mucosa of patients with Crohn's disease (CD) included in cohort 1 based on microarray analysis. (A) Venn diagram showing the intersection of differentially expressed genes in intestinal colonic mucosa from patients with CD versus controls with non-inflammatory bowel disease (non-IBD). Differentially expressed genes in patients with active CD without anti-tumour necrosis factor α (anti-TNFα; n=10), patients with active CD with anti-TNFα (non-responders, n=5) and patients with inactive CD with anti-TNFα (responders, n=7), compared with non-IBD controls (n=17) are shown. The number of differentially expressed genes (DEGs) is shown in black. Asterisks highlight the subset of most relevant genes. (B, C) Heatmap representation of differentially expressed genes in colonic mucosa according to anti-TNFα response. Each row shows one individual gene and each column an experimental sample. Differentially upregulated genes are shown in red and downregulated genes are shown in green. Unsupervised hierarchical cluster method using Pearson distance and average linkage method was applied for gene and sample classification. Samples from non-IBD controls (black, n=17), patients with active CD without anti-TNFα (red, n=10), patients with active CD with anti-TNFα (non-responders; green, n=5) and patients with inactive CD with anti-TNFα (responders; blue, n=7) are shown. (B) Heatmap representation of microarray data from genes whose expression was significantly regulated in all patients with active CD, independently of anti-TNFα administration. (C) Heatmap representation of microarray data of genes modulated by anti-TNFα both in responders and non-responders.
Despite the common gene signature seen in patients with active disease receiving (or not) anti-TNFα, our analysis identified a set of genes that were significantly regulated by TNFα blockade, regardless of endoscopic remission. Indeed, 612 of the 1310 genes that were regulated in patients with active CD returned to non-IBD control levels in non-responders (patients with persistent endoscopic activity despite anti-TNFα treatment) (figure 3A, double asterisk). Of those, 38 genes (figure 3C) also fulfilled the following criteria: (1) they were equally expressed in CD responders, non-responders and non-IBD controls; and (2) their expression in anti-TNFα treated (responders or non-responders) versus those with active CD but without anti-TNFα therapy differed significantly. This set of genes was regulated by anti-TNFα, even in non-responders and included IL6, CD69, GPR183, MMP9, among others (table 3).
Relative changes (fold-change) in the transcription of those genes that were significantly modulated by anti-tumour necrosis factor α (anti-TNFα) treatment in non-responders
IL6 gene expression is significantly regulated by anti-TNFα therapy regardless of response
IPA for the subset of 38 genes regulated by anti-TNFα (table 3) identified a connecting network involving many of the molecules significantly regulated in our dataset. This included IL-6, which was the most highly downregulated gene in anti-TNFα-treated patients regardless of mucosal healing (figure 4A). In order to validate our findings, we determined the expression of IL6 transcripts in an independent cohort of patients via real-time RT-PCR (cohort 2, table 1). The transcription patterns of IL1B and S100A8, which in contrast to IL6 correlated with the presence of inflammation in anti-TNFα non-responders, were also analysed (figure 4B). As shown in figure 4B, the differences apparent in microarray analysis in cohort 1 were reproduced by real-time RT-PCR in cohort 2: IL1B and S100A8 were upregulated in patients with active CD (whether or not they were receiving anti-TNFα treatment) compared with patients in remission (responders), while IL6 expression was significantly increased only in those patients with active CD not receiving anti-TNFα. In order to determine whether the genes associated with anti-TNF response in non-responders were specific to this treatment, we analysed the expression of IL6, IL1B and S100A8 in patients receiving azathioprine alone and compared responders with non-responders (presenting mucosal lesions after 4 months’ treatment with azathioprine). All three genes were significantly downregulated in responders compared with non-responders, but remain elevated in non-responders (see online supplementary figure S1), suggesting that the downregulation of IL6 gene expression is specifically induced by blockade of TNFα.

Transcription of IL6 is significantly regulated by anti-tumour necrosis factor α (anti-TNFα) therapy independently of response to treatment. (A) Network showing the interactions between the subset of molecules identified as TNFα-regulated genes in responders and non-responders based on the Ingenuity Knowledge Base Ingenuity Pathways Analysis (IPA). Downregulated molecules are shown in green and those upregulated in red. Colour intensity correlates with the observed change; extreme colour indicates a higher observed change and vice versa. (B) Box plot representation of transcriptional expression of IL6, IL1B and S100A8 as assessed by microarray (cohort 1, Table 1) and real-time RT-PCR validation (cohort 2, Table 1). Statistical significance between patients with active Crohn's disease (CD) with and without anti-TNFα therapy and patients with inactive CD with anti-TNFα are shown (*p≤0.05). Non-IBD, controls with non-inflammatory bowel disease.
Pathway analysis of persistently activated genes in non-responders identifies IL-1B and IL-17A as potential alternative targets
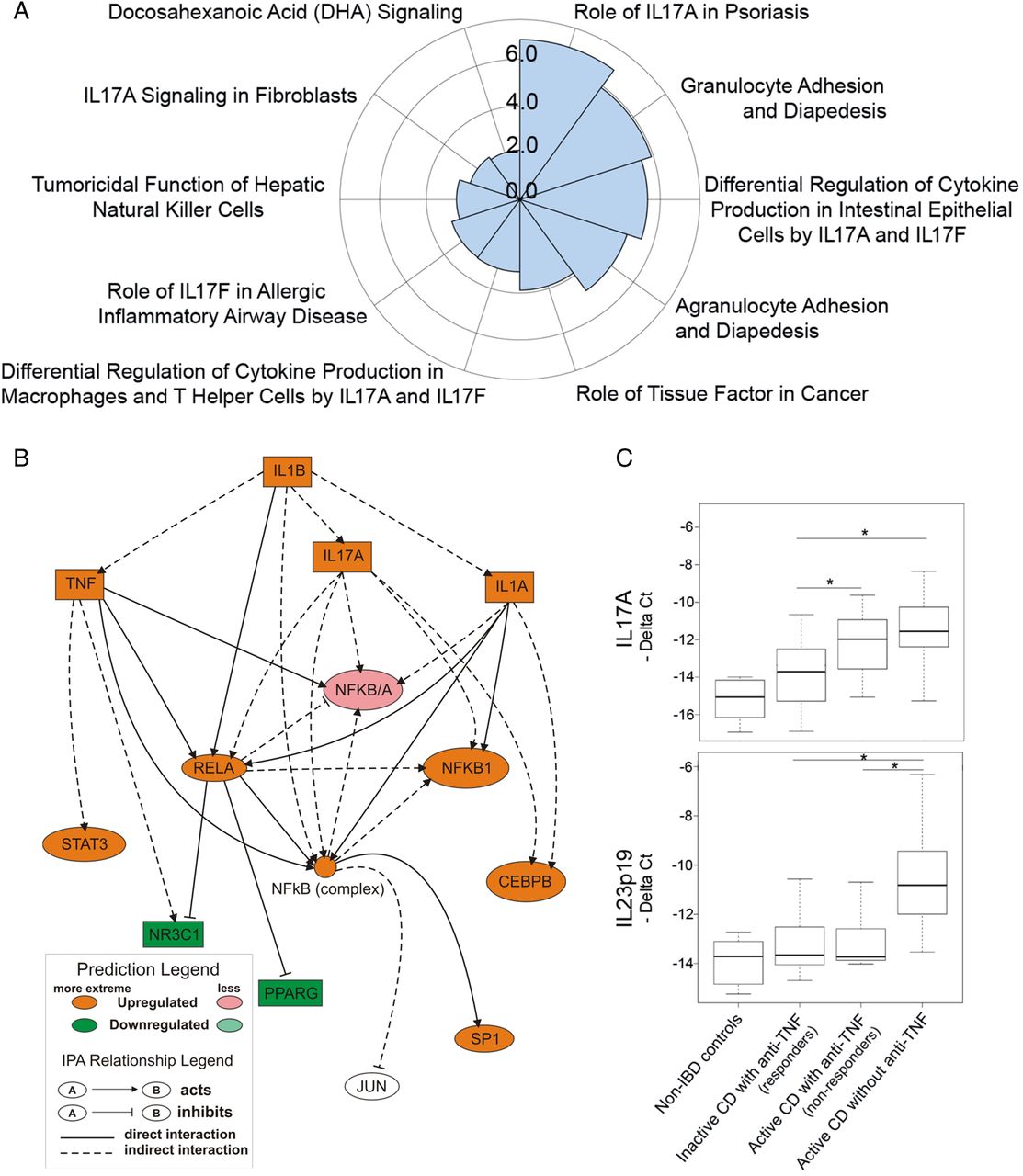
To determine the mechanisms that maintain mucosal inflammation in anti-TNFα non-responders, we performed IPA of the 170 genes that remained deregulated in these patients (see online supplementary table S2). Analysis of the involved canonical pathways identified several IL-17A-related pathways as significantly regulated in non-responding patients, suggesting the presence of a dominant IL-17A-driven response that was not sensitive to an anti-TNFα blockade in non-responders (figure 5A). Moreover, the mechanistic networks for candidate upstream regulators were assessed. Figure 5B shows a network encompassing two of the most relevant upstream regulators identified, IL1B and IL17A, both of which were predicted to be upregulated genes in non-responders. The computational predicted behaviour of IL1B was confirmed by our microarray data and validated by real-time RT-PCR (see online supplementary table S2 and figure 4B). Expression of IL17A was not detectable in our microarray analysis, owing to the probe's low signal threshold. We used real-time RT-PCR analysis to determine the levels of IL17A transcription in all four subject groups (figure 5C). IL17A was significantly upregulated in patients with active CD, independently of treatment, compared with patients in remission (responders). Therefore, both IL-1B and IL17A remained significantly upregulated in patients refractory to anti-TNF α therapy, which is in agreement with the Th17 signature identified in our canonical pathway analysis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Pathway analysis of persistently activated genes in non-responders identifies IL-1B and IL-17A as potential alternative targets. (A) Polar graph showing the top 10 canonical pathways associated with persistently activated genes in refractory patients identified by Ingenuity Pathways Analysis (IPA). Results are shown graphically as a negative logarithm of the probability score (the most statistically significant pathways have the highest value in the graph). (B) Representation of mechanistic network encompassing two plausible upstream regulators found by IPA analysis. The prediction molecule for downregulation is shown in green and that for upregulation in red. Colour intensity correlates with the predicted change; extreme colour indicates a higher expected change and vice versa. (C) Box plot representation of transcriptional expression patterns of IL17A and IL23p19 as assessed by real-time RT-PCR. Statistical significance between patients with active Crohn's disease (CD) with and without anti-tumour necrosis factor α (anti-TNFα) therapy and patients with inactive CD with anti-TNFα are shown (*p≤0.05).
Finally, three additional target genes, IL23p19, IL12p35 and IL17F were assessed owing to their clinical relevance as potential targets for modulating mucosal damage in this group of patients with refractory disease. Interestingly, IL23p19 appeared to be downregulated in anti-TNFα-treated patients regardless of their endoscopic response (figure 5C), while IL12p35 expression remained low in all samples (cycle threshold (Ct) range 29.4–35.3) and no significant differences between CD groups were noted (data not shown). Similarly, IL17F expression was low in healthy controls (Ct range 34.4–37) and while it was significantly upregulated in patients with CD the Ct values remained low (Ct range 30.9–36.03) and no differences were detected among CD groups (data not shown).
Discussion
In this study we used whole-genome transcriptional analysis of the colonic mucosa to examine previously undescribed mechanisms related to failure of anti-TNFα therapy in CD. Our goal was to reveal new targets that would benefit patients with refractory disease. While transcriptional expression analysis of intestinal biopsy specimens in patients with IBD has been extensively used to identify biomarkers, treatment response predictors and new disease mechanisms,13–19 to the best of our knowledge no information is available on the whole-genome transcriptional signature of anti-TNFα non-responders following treatment failure.
Our study first identified the transcriptional changes that were associated with anti-TNFα-induced mucosal healing in CD. Next, we compared the changes achieved in anti-TNFα responders with those in non-responders. Using this approach, we could discriminate between inflammatory mechanisms that are dependent on TNFα signalling and those that are independent. We hypothesise that those genes that remain deregulated in anti-TNFα non-responders might represent alternative targets in this important subset of refractory subjects. In contrast, disease-independent TNFα-regulated genes (those that are sensitive to anti-TNFα therapy even in non-responders) may support the presence of non-dominant disease mechanisms in this patient group. Our data suggest that therapeutic interventions directed against these targets are not likely to afford a significant benefit to these patients and may therefore not represent promising alternatives for non-responders.
Among the genes identified as being regulated upon TNFα blockade (even in unresponsive patients with persistent mucosal lesions) were IL6, IL23p19, MMP9, urokinase PLAU and cell-activation markers (ie, CD69, CD83 and VCAM1), all of which are known mediators of inflammation. Interestingly, the IL23R genotype has been shown to influence the response to anti-TNFα treatment.20 Also, the anti-IL-6 receptor antibody tocilizumab21 has shown efficacy in both inducing and maintaining remission in CD; whether IL-6 blockade might confer additional benefits to anti-TNFα non-responders, however, has not yet been examined. It is not surprising that many of the genes regulated by anti-TNFα antibodies, both in responders and non-responders, are known to be dependent upon TNFα signalling (ie, the apoptosis-inducing p53 target PMAIP1),22 the TNFα-induced proteins TNFAIP3 and TNFAIP8, and the NF-κB-dependent TNFα-induced genes CD69,23 ,24 MMP9,25 VCAM1,26 PLAU (urokinase-type plasminogen activator)27 and CD83.28 The regulation of this gene set by anti-TNFα antibodies, even in non-responders, suggests that the lack of response to anti-TNFα in these patients may not reflect the absence of biological activity of the administered antibody. In addition, our data support the possibility of a redundant role of these mediators in maintaining inflammation, at least in patients who fail to respond to anti-TNFα therapy.
In agreement with previous reports, we here demonstrate that anti-TNFα-induced mucosal healing is associated with a significant modulation of a large number of genes in patients with CD,13 ,14 ,18 ,29 including genes related to the Th17 pathway30–33 and genes such as PRDM1 with known genetic variants associated with CD susceptibility.34 Moreover, we show a correlation of expression of this gene group with both endoscopic and radiological activity, as measured by global CDEIS and MaRIA score.35 This suggests they might potentially serve as biomarkers of response to anti-TNFα.36 Genes such as IL1B, S100A8, S100A9, CXCL2, CXCL6, S100A12, SLC64A14, FCGR3B (CD16b) and the anti-bacterial genes DMBT1 and IDO1, regulated by IL2737 or IL17,38 as well as IL17A itself, were significantly upregulated in patients with active CD, including anti-TNFα non-responders. We propose that this set of genes may drive inflammation, at least in non-responders, independently of TNFα signalling and therefore their targeting might benefit anti-TNFα non-responders. IL17A has recently been targeted in patients with CD, but conferred no apparent benefits and was associated with a high rate of adverse effects.39 Although anti-IL-1B antibodies are successful in treating other inflammatory conditions, they remain to be tested in IBD.40 ,41
The main limitation of our observational and exploratory study is the relatively small cohort sizes, and further microarray-based studies will be necessary to confirm our findings. Nonetheless, our results put forward the new concept that transcriptional analysis in patients with persistent inflammation despite available treatments might be an unexploited tool for identifying alternative treatments.
Although one might interpret some of the changes in the mucosal gene expression of ‘non-responders’ as partial changes, reflecting incomplete or insufficient effect of the drug, all non-responders presented mucosal lesions resistant to anti-TNF therapy. We show that those lesions share a common transcriptional signature with the inflamed mucosa from patients not receiving anti-TNFα. Moreover, they regulate a specific set of 38 genes after anti-TNFα treatment despite their failure to heal the mucosa. We believe that this reflects a specific pathway being targeted by anti-TNFα that operates independently of complete remission and not simply a partial response.
In summary, we show that anti-TNFα antibodies, even in patients who do not improve with treatment, regulate several TNFα-dependent genes. Interestingly, regulation of these well-known mediators of inflammation and markers of cell activation does not necessarily result in the healing of mucosal lesions in all patients. Whether this reflects the presence of alternative inflammatory pathways that maintain mucosal lesions and prevent tissue recovery in anti-TNFα non-responders cannot be concluded from our results. Importantly, we demonstrate that IL17A and IL1B may be persistent inflammatory mediators, supporting the presence of TNFα-resistant inflammation in non-responders, whereas IL6 is profoundly downregulated by anti-TNFα antibody therapy, even in non-responders. Our new results may be instrumental in guiding the choice of treatments to be used in patients who fail to respond to anti-TNFα treatment.
Acknowledgments
We thank the endoscopy department at Hospital Clinic Barcelona for providing us with the samples required to conduct this study and our patients for their selfless participation. We are indebted to Joe Moore for editorial assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figure 1
- Data supplement 3 - Online table 1
- Data supplement 4 - Online table 2
Footnotes
-
Contributors AS, RK, HP and JP designed the study; MCM, IO and ER recruited patients; IO and ER assessed clinical disease activity; RFL, NP, ID and ME performed the experiments; NP and JJL carried out bioinformatic and biostatistic analysis; and AS, NP, RFL and JP wrote the manuscript.
-
Funding This work was supported by Hoffmann-La Roche Pharmaceuticals (Nutley, New Jersey, USA) and grant BFU2012-35999 to AS from the Spanish Ministry of Economy. RFL was supported by a research fellowship from CAPES-Brazil.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Ethics committee, Hospital Clinic of Barcelona.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement Microarray raw data from samples included in this study have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE52746; private access until manuscript publication (reviewer's link: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=kdobkkgmtdujvsj&acc=GSE52746).