Article Text

Abstract
Background & aims Inflammatory bowel disease (IBD) is characterised by chronic intestinal inflammation, resulting from dysregulation of the mucosal immune system and compromised intestinal epithelial barrier function. The bile salt, nuclear farnesoid X receptor (FXR), was recently implicated in intestinal antibacterial defence and barrier function. The aim of this study was to investigate the therapeutic potential of FXR agonists in the treatment of intestinal inflammation in complementary in vivo and in vitro models.
Methods Colitis was induced in wild-type (WT) and Fxr-null mice using dextran sodium sulfate, and in WT mice using trinitrobenzenesulfonic acid. Mice were treated with vehicle or the FXR agonist INT-747, and colitis symptoms were assessed daily. Epithelial permeability assays and cytokine expression analysis were conducted in mouse colon and enterocyte-like cells (Caco-2/HT29) treated with medium or INT-747. Inflammatory cytokine secretion was determined by ELISA in various human immune cell types.
Results INT-747-treated WT mice are protected from DSS- and TNBS-induced colitis, as shown by significant reduction of body weight loss, epithelial permeability, rectal bleeding, colonic shortening, ulceration, inflammatory cell infiltration and goblet cell loss. Furthermore, Fxr activation in intestines of WT mice and differentiated enterocyte-like cells downregulates expression of key proinflammatory cytokines and preserves epithelial barrier function. INT-747 significantly decreases tumour necrosis factor α secretion in activated human peripheral blood mononuclear cells, purified CD14 monocytes and dendritic cells, as well as in lamina propria mononuclear cells from patients with IBD.
Conclusions FXR activation prevents chemically induced intestinal inflammation, with improvement of colitis symptoms, inhibition of epithelial permeability, and reduced goblet cell loss. Furthermore, FXR activation inhibits proinflammatory cytokine production in vivo in the mouse colonic mucosa, and ex vivo in different immune cell populations. The findings provide a rationale to explore FXR agonists as a novel therapeutic strategy for IBD.
- FXR
- DSS-induced IBD
- TNBS-induced IBD
- intestinal permeability
- immune modulation
- epithelial permeability
- gut inflammation
- IBD models
- intestinal barrier function
- mucosal barrier
Statistics from Altmetric.com
- FXR
- DSS-induced IBD
- TNBS-induced IBD
- intestinal permeability
- immune modulation
- epithelial permeability
- gut inflammation
- IBD models
- intestinal barrier function
- mucosal barrier
Significance of this study
What is already known about this subject?
Inflammatory bowel disease (IBD) is characterised by chronic intestinal inflammation, resulting from dysregulation of the mucosal immune system and compromised intestinal epithelial barrier function.
Bile duct obstruction in humans and mice is associated with mucosal injury, bacterial overgrowth and translocation. Oral administration of bile salts or farnesoid X receptor (FXR) agonists counteracts these deleterious effects.
FXR activation protects against inflammation presumably by repression of NF-κB signalling in mice.
FXR activation counter-regulates inflammatory cytokine expression in activated murine and human immortalised immune cells.
What are the new findings?
FXR activation ameliorates intestinal inflammation at multiple levels.
Dextran sodium sulfate- and trinitrobenzenesulfonic acid-induced colitis in mice results in significant goblet cell loss, and FXR activation partially protects from the decrease in goblet cells.
FXR activation preserves the integrity of the intestinal epithelial barrier.
FXR activation counteracts NF-κB-mediated immune responses in enterocytes in vitro.
FXR activation inhibits inflammatory signalling in various primary human immune cell types (peripheral blood mononuclear cells, CD14 monocytes and monocyte-derived dendritic cells). Moreover, the anti-inflammatory properties of FXR activation are confirmed ex vivo in lamina propria mononuclear cells from patients with IBD.
How might it impact on clinical practice in the foreseeable future?
This multi-level protection against intestinal inflammation provides a rationale to explore FXR agonists as a novel therapeutic strategy for IBD in humans.
Introduction
Inflammatory bowel disease (IBD), including Crohn disease (CD) and ulcerative colitis (UC), is characterised by chronic intestinal inflammation, with potentially severe complications and even mortality.1 Although the exact aetiology is unclear, it is thought to result from a combination of dysregulation of the mucosal immune system and compromised intestinal epithelial barrier function in genetically predisposed individuals.2 Several genes associated with IBD are involved in antibacterial defence (eg, NOD2, ATG16L1, cathelicidin, defensins) as well as barrier function (eg, PTPsigma, MAGI2, myosin IXB and E-cadherin).3–6 Current treatment options for IBD are mainly aimed at suppressing the immune response. However, although they are reasonably effective, significant side effects and treatment failures can occur, highlighting the need for novel treatment options in IBD.
The intestinal epithelial barrier protects the host by preventing external antigens and micro-organisms from entering the body.7 This physical barrier consists of enterocytes tightly connected via intercellular junctions. Enterocytes also secrete cytokines and chemokines, which trigger the inflammatory response, as a second line of defence against luminal contents. In addition, mucin-secreting goblet cells form the mucus layer that protects the mucosal surface from antigens and helps to maintain the intestinal barrier function. Patients with IBD with clinically and endoscopically significant colitis display various degrees of goblet cell loss.8 The epithelial barrier is often already compromised in early stages of the disease, leading to bacterial translocation and inflammation.9 10
Dysregulation of the immune response to intestinal bacteria in patients with IBD occurs because of a shift of balance from secretion of anti-inflammatory mediators towards proinflammatory molecules. Activation of the nuclear transcription factor κB (NF-κB) was identified as a key factor in this shift, resulting in strongly enhanced expression of proinflammatory genes and recruitment of excess inflammatory cells to the intestinal wall.
Recently, the nuclear farnesoid X receptor (FXR) has been implicated in immune modulation and barrier function in the intestine.11 FXR is activated by bile salts and regulates transcription of genes involved in bile salt synthesis, transport and metabolism in the liver and intestine by binding FXR responsive elements in promoters of target genes as a heterodimer with retinoid X receptor (RXR).12 In the rodent bile duct ligation model and in humans with bile duct obstruction, bile salt deficiency in the intestine is associated with mucosal injury, bacterial overgrowth and translocation. Strikingly, oral administration of bile salts or FXR agonists counteracted these deleterious effects.13–16 Fxr-null mice exhibited compromised intestinal integrity at baseline, with further deterioration after bile duct ligation, which was not prevented by FXR agonist treatment.15 Recently, FXR activation was also shown to protect against hepatic inflammation presumably by repression of NF-κB signalling in mice.17 In addition, FXR is expressed and counter-regulates inflammatory cytokine expression in activated immune cells.18 19 Thus, immune cell modulation by FXR activation may be responsible for the improvement in intestinal inflammation.11
Here, we have applied an integrated in vitro and in vivo approach to investigate the therapeutic potential of FXR agonists for the treatment of intestinal inflammation. We specifically focused on the crosstalk between the epithelial barrier and immune modulation by enterocytes as well as immune cells. We show that treatment with FXR agonists ameliorates intestinal permeability in vivo and in vitro, and provide evidence that FXR activation counteracts proinflammatory cytokine expression and secretion by enterocytes and by different human immune cell types. The anti-inflammatory properties of the selective FXR agonist, INT-747, were also observed in ex vivo lamina propria mononuclear cells (LPMCs) from patients with IBD.
Methods
Animals
Wild-type (WT) C57BL/6J mice (9–12 weeks old) were obtained from Charles River Laboratories (Calco (Lecco), Italy). Pure strain C57BL/6J Fxr-null mice (Dr D J Mangelsdorf (Southwestern Medical Center, Dallas, Texas, USA) were kindly provided by Dr Frank Gonzalez (NIH, Bethesda, Maryland, USA). Mice were fed ad libitum and housed in a temperature- and light-controlled room. All experiments were approved by the ethics committee of the Consorzio Mario Negri Sud.
Pharmacological activation of FXR and colitis induction
Pharmacological activation of FXR was accomplished by daily oral gavage with 5 mg/kg/day INT-747 (6-ethylchenodeoxycholic acid; Intercept Pharmaceuticals Inc, New York, New York, USA)20 or vehicle (1% methyl cellulose) from 3 days before chemically induced colitis treatments, and continued until the end of the experiments. Two well-established models for colitis were used. First, colitis was induced by administration of 2.5% (w/v) dextran sodium sulfate (DSS; molecular mass 36–50 kDa; MP Biochemicals Inc, Amsterdam, The Netherlands) in drinking water for 7 or 10 days (n=8–10 mice per group). Second, colitis was induced by two rectal administrations (rectal silicon catheter 3.5 French; Instech Solomon Scientific, San Antonio, Texas, USA) of 1.5 mg trinitrobenzenesulfonic acid (TNBS) in 40% ethanol, with a time interval of 7 days. Mice (n=5–8 mice per group) were killed 48 h after the second TNBS administration. Daily changes in body weight were assessed. Rectal bleeding was scored on a scale from 0 to 5, indicating no (0) to very severe 5 rectal bleeding. For intestinal permeability assays, mice were killed after 7 days of DSS treatment or 48 h after the second TNBS administration. Ilea and colons were snap-frozen or fixed in 10% formalin (24 h) and embedded in paraffin.
mRNA extraction and quantitative real time qRT-PCR analysis
RNA was isolated from ileum and colon of mice using RNeasy Micro kit (Qiagen, Venlo, The Netherlands). RNA was isolated from HT29 and Caco-2 cells using TRIzol reagent. cDNA was generated from 500 ng total RNA using SuperScript II Reverse Transcriptase (Invitrogen, Carlsbad, California, USA). qRT-PCR analysis was carried out using SYBR green PCR mastermix and analysed on a MyIQ real time PCR cycler (BioRad, Veenendaal, The Netherlands). Values were normalised to glyceraldehyde-3-phosphate dehydrogenase. Primers are listed in supplementary table 1 (online).
Bile salt measurements
Conjugated bile salt species were analysed by high-performance liquid chromatography21 using a C-18 Waters Bondapack 10 μm column (Waters, Milford, Massachusetts, USA). Methanol/phosphate buffer was used as the eluent (pH 5.2, flow rate 1 ml/min).
Histology
Distal colon sections (5 μm) were stained with H&E. Histopathological scoring was performed by an experienced pathologist blinded to the experimental conditions, using an established semiquantitative score ranging from 0 to 6 based on infiltration of inflammatory cells and epithelial damage (1=few inflammatory cells, no epithelial degeneration; 2=mild inflammation, few signs of epithelial degeneration; 3=moderate inflammation, few epithelial ulcerations; 4=moderate to severe inflammation, ulcerations in 25–50% of the tissue section; 5=moderate to severe inflammation, large ulcerations in >50% of the tissue section; 6=severe inflammation and ulcerations of >75% of the tissue section).22 Depletion of goblet cells was scored using a scoring index from 0 to 4 (0=no depletion; 1=0–10% depletion; 2=10–25% depletion; 3=25–50% depletion; 4=50–100% depletion).
In vivo intestinal permeability assay
Intestinal permeability was examined in mice after 7 days of DSS treatment or 48 h after the second TNBS administration, as described previously.23 Results were normalised to WT mice, which did not receive any treatment (n=3). Mice were gavaged with 0.6 mg/g body weight of fluorescein isothiocyanate (FITC)-conjugated dextran (Sigma, S Louis, Missouri, USA; molecular mass 3–5 kDa) for 4 h. Blood was collected, and FITC concentrations were measured in plasma (Fluorimeter Pharos FX; BioRad, Hercules, California, USA).
Permeability and lactate dehydrogenase (LDH) release assays in Caco-2 cell monolayers
Caco-2 cells were grown on collagen-treated polytetrafluoroethylene (PTFE) Transwell filter inserts (pore size=0.4 μm; Corning Inc, Big Flats, New York, USA), differentiated for 14 days, and incubated with or without 2.5% DSS for 10 days, in the presence or absence of 1 μM FXR synthetic ligand GW4064 (started 3 days before DSS treatment). At day 6, 7, 8, 9 and 10, medium containing 100 μM cell-impermeable Lucifer Yellow (Sigma) was added to the apical compartment. After 1.5 h, fluorescence was measured in basolateral medium (FLUOstar Galaxy microplate reader; BMG LabTech, Offenburg, Germany). The flux of the Lucifer Yellow into the basolateral compartment was calculated as the percentage of total fluorescence applied to the apical compartment. At day 8 of DSS treatment, medium from the apical compartment was harvested for assessment of LDH release (cytotoxicity detection kit; Roche Applied Science, Almere, The Netherlands). Results were normalised to cultures treated with 1% Triton X100 (Sigma) for 10 min. At day 8, representative phase-contrast images of the transwells were recorded.
Cell cultures
HEK293T, HT29 and Caco-2 cells were grown in Dulbecco modified Eagle's medium (GlutaMax), supplemented with 10% fetal calf serum and 1% penicillin/streptomycin (Gibco BRL, Breda, The Netherlands). Caco-2 cells were co-treated with 1% non-essential amino acids. To obtain differentiated HT29 and Caco-2 cells, they were maintained in culture for 7 and 14 days after confluency, respectively.
Reporter assays
HEK293T cells were co-transfected with a reporter construct containing the responsive element for NF-κB (pGL3-2κB luc) and pcDNA or pcDNA-FXR or pcDNA-FXRW469A (ligand-binding domain mutant)24 together with pcDNA-RXRα using the calcium phosphate method. At 24 h after transfection, cells were incubated with vehicle (dimethyl sulfoxide) (DMSO) and 1 μM GW4064 for 24 h in the presence or absence of tumour necrosis factor (TNF)α (500 U/ml). Cells were lysed and Firefly and Renilla luciferase activity were measured (Promega Dual-Luciferase Reporter Assay System; Promega, Madison, Wisconsin, USA) with the Centro LB 960 luminometer (Berthold Technologies, Vilvoorde, Belgium).
Purification and culture of human immune cells
Cells were cultured in RPMI 1640 GlutaMax culture medium supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Charles River, Lecco, Italy), 1% non-essential amino acids, 0.5 mg/ml gentamicin and 1 mM sodium pyruvate. Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats of healthy subjects by Ficoll gradient (BioChrome AG, Berlin, Germany), and monocytes were purified from PBMCs by positive selection with CD14 beads (Milteny Biotec, Bergish Gladbach, Germany). PBMCs or monocytes were cultured, as previously described,25 in the presence of 100 ng/ml lipopolysaccharide (LPS) from Escherichia coli 0111:B4 (Sigma-Aldrich, Milano, Italy), with or without 10 mM INT-747. Supernatants were harvested after 24 h and stored at −80°C until analysis.
Monocyte-derived dendritic cells were differentiated from purified monocytes26 and cultured in complete medium containing 10 ng/ml recombinant human granulocyte-macrophage colony stimulating factor (GM-CSF) and 10 ng/ml recombinant human interleukin (IL)-4 (PharMingen, San Diego,California, USA) in the presence or absence of 10 mM INT-747. After 7 days, cells were harvested and either stimulated for 24 h with 100 ng/ml LPS or immediately stained with monoclonal antibodies, anti-CD1a phycoerythrin (PE) (HI149) and anti-CD14 FITC (M5E2) (Pharmingen). Flow cytometric analysis was performed with a FACScalibur flow cytometer (Becton Dickinson, Mountain View, California, USA). LPMCs were isolated from colonic or ileal biopsy specimens from two patients with active UC and two patients with active CD, as previously described.27 One patient with UC was receiving 5-aminosalicylic acid treatment, and the second was taking azathioprine. One patient with CD was taking no medication, and the second was taking azathioprine. Patients gave their informed consent, and the study was approved by the local ethics committee.
Enriched lamina propria leucocytes were cultured in complete medium supplemented with 100 IU/ml penicillin, 0.1 mg/ml streptomycin and 0.5 μg/ml amphotericin B in the presence of 1 μg/ml coated anti-CD2 and soluble anti-CD28 (BD Biosciences, Milano, Italy). Cells were treated with vehicle or 10 mM INT-747, and supernatants were harvested after 24 h or 72 h as specified in the figure legends and stored at −80°C until analysis.
ELISAs for human interferon γ (IFNγ), TNFα and IL-6 were performed using monoclonal antibody pairs and standards provided by BD OptEIA Human ELISA set (BD Biosciences), and myeloperoxidase (HBT, Uden, The Netherlands), according to the manufacturers' procedures.
Statistical analysis
Results are expressed as mean±SEM or mean±SD as indicated in the figure legends. Statistical significance was determined by the Student t test or analysis of variance with Bonferroni post hoc test as appropriate. All statistical calculations were performed with SPSS 15.0 software. Two-sided p values (p<0.05, p<0.01, p<0.001) are considered significant.
Results
Oral gavage with INT-747 activates Fxr in ileum and colon and enters the enterohepatic circulation
To confirm that the dose of INT-747 applied activates FXR in vivo in the intestine, FXR target gene expression in the ileum and colon was assayed. In WT mice, Fxr mRNA expression did not change on INT-747 treatment. However, FXR target genes were induced both in ileum (small heterodimer partner Shp, 52-fold; fibroblast growth factor Fgf15, 4-fold) and colon (Shp, 19-fold). Fxr-null mice expressed very low levels of Shp and Fgf15, which did not change on INT-747 treatment (online supplementary figure 1).
Taurine-conjugated INT-747 is enriched in bile of WT (9.8±0.8%) and Fxr-null (5.3±0.8%) mice after oral gavage with INT-747 for 13 days, indicating that INT-747 enters the enterohepatic circulation. In addition, the ratio between tauro-conjugated cholic acid and β-muricholic acid is decreased by INT-747 treatment (1.16±0.22 vs 0.57±0.11), suggesting that Cyp7a1-mediated hepatic cholic acid synthesis is repressed by FXR activation. Together, these results indicate that INT-747 is retained in the enterohepatic circulation with efficient activation of FXR and its target genes.
Fxr activation by INT-747 decreases symptoms of DSS-induced colitis
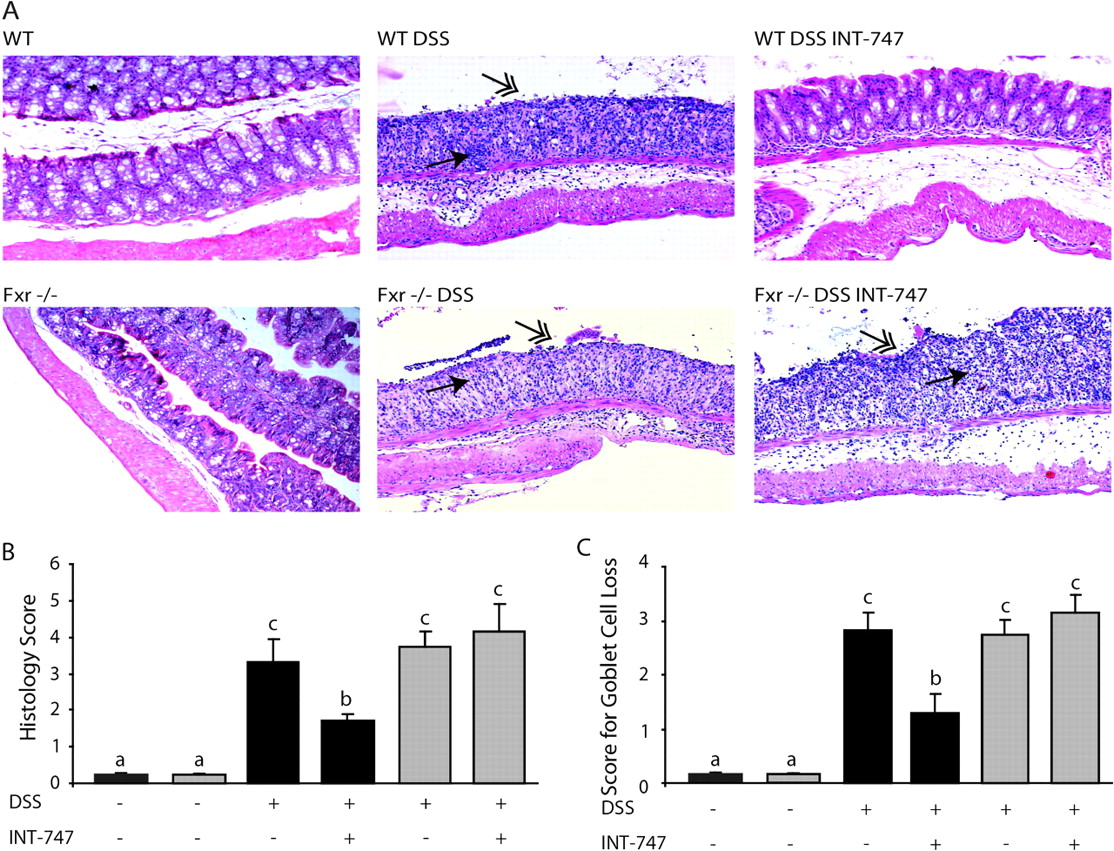
We investigated whether INT-747-mediated FXR activation confers protection against DSS-induced colitis. INT-747 treatment significantly reduced body weight loss (figure 1A) and rectal bleeding scores (figure 1B) and prevented colonic shortening (figure 1C) in WT mice (normal colon length 6.5–7 cm; data not shown). In addition, histological analysis showed that DSS-induced colitis was associated with complete disruption of the epithelial layer and acute inflammatory infiltrates in WT and Fxr-null mice. In sharp contrast, INT-747-treated WT mice showed significantly less morphological alteration and decreased inflammatory infiltrates. In addition, goblet cell loss due to DSS-induced inflammation was significantly less in INT-747-treated WT mice (figure 2). INT-747 had no effect on colitis symptoms and histology in Fxr-null mice.

INT-747-dependent Fxr activation confers protection against clinical symptoms of DSS-induced colitis. (A) Percentage of initial body weight during DSS treatment and (B) rectal bleeding score; analysis of variance with Bonferroni post hoc test was performed for each time point; significant differences (p<0.05) are indicated by different letters. (C) Colon length (cm) of wild-type (WT; black bars) and Fxr-null mice (grey bars); Student t test, *p<0.05 compared with WT DSS vehicle-treated mice. (WT -; Fxr-null x; WT DSS ◆; WT DSS + INT-747 ●; Fxr-null DSS ▲; Fxr-null DSS + INT-747 ■).

INT-747-dependent Fxr activation ameliorates histological characteristics of inflammation. (A) H&E-stained colonic sections. Some dextran sodium sulfate (DSS)-treated wild-type (WT) mice that received INT-747 were almost completely protected from DSS-induced inflammation (upper left panel). Single-headed arrows point to inflammatory infiltrates and double-headed arrows to epithelial degeneration. Magnification 100×. (B) Histology score. (C) Score for goblet cell loss. Each bar represents mean±SEM. Analysis of variance with Bonferroni post hoc test was performed; significant differences (p<0.05) are indicated by different letters.
In a separate experiment, in which WT mice were treated with DSS for 5 days and were resumed on water until day 10, body weight, rectal bleeding, stool consistency and colon length approached normal values much more quickly in INT-747-treated than in vehicle-treated mice, indicating the capacity of this compound to reduce symptom duration (online supplementary figure 2A–D). Together, these data indicate that treatment with INT-747 results in significant amelioration of DSS-induced IBD in WT, but not Fxr-null, mice.
Fxr activation decreases DSS-induced intestinal permeability in vivo
We next investigated whether Fxr activation affects DSS-induced epithelial permeability. Plasma levels of FITC-conjugated dextran were markedly increased in WT and Fxr-null mice in which DSS-mediated colitis was induced (figure 3A). INT-747 treatment almost completely abolished DSS-induced permeability in WT, but not Fxr-null, mice. Also, in animals without DSS treatment, intestinal permeability was much lower in WT than Fxr-null mice (online supplementary figure 3). These data indicate that INT-747-dependent FXR activation preserves the integrity of the intestinal epithelial barrier.

FXR activation preserves the integrity of the intestinal epithelial barrier in vivo. (A) In vivo intestinal permeability measurement after 7 days of dextran sodium sulfate (DSS) of vehicle- or INT-747-treated wild-type (WT) and Fxr-null mice. Each data point represents mean±SEM. Student t test, **p<0.01 compared with WT vehicle-treated mice. (B) Lucifer Yellow permeability measured in differentiated Caco-2 cell monolayers (0% DSS ◆; 0% DSS GW4064 ●; 2.5% DSS ■; 2.5% DSS GW4064 ▲); analysis of variance with Bonferroni post hoc test was performed for each time point; significant differences (p<0.05) are indicated by different letters. (C) Bright field microscopy pictures and (D) LDH assay after 8 days of DSS treatment. Caco-2 cells incubated with 1% Triton (white bar), dimethyl sulfoxide (DMSO; light grey bar) or GW4064 (black bar).
FXR activation protects against DSS-induced permeability in Caco-2 cell monolayers
To study the effects of FXR activation on intestinal permeability in vitro, we studied apical to basolateral flux of the cell-impermeable Lucifer Yellow compound as a measure of permeability of differentiated Caco-2 cells. Caco-2 cells are enterocyte-like cells which have previously been described in permeability assays.28 29 FXR is only expressed and functional in fully differentiated Caco-2 cells, comparable to in vivo expression30 (online supplementary figure 4A,B). Cells treated with 2.5% DSS showed increased permeability of the monolayer starting between days 7 and 8. However, permeability was markedly decreased on co-treatment with the synthetic FXR agonist GW4064 (figure 3B), suggesting that FXR activity confers protection against DSS-induced monolayer permeability. In line with this finding, the typical Caco-2 cell monolayer morphology changed dramatically at day 8: cells detached from each other and the transwell, leaving gaps in the monolayer (figure 3C). Medium taken from the same experiment (day 8) did not show increased LDH release in Caco-2 cells on DSS treatment compared with untreated cells at the same time point (figure 3D), indicating that the DSS-induced permeability is not caused by cell damage.
Fxr activation inhibits inflammatory gene expression and promotes antibacterial gene expression in DSS-induced colitis
Since IBD in humans is thought to result from both compromised intestinal epithelial barrier function and dysregulation of the mucosal immune system, we investigated whether FXR modulates inflammatory gene expression in DSS-induced colitis. In DSS-treated WT mice, INT-747 significantly decreased colonic mRNA expression of the proinflammatory genes Il-1β, Il-6 and macrophage attractant protein-1 (Mcp-1) (figure 4A). These data are consistent with the significantly reduced colonic protein concentrations of Il-6, Mcp-1 and myeloperoxidase in INT-747-treated mice (online supplementary figure 2E–G).

Fxr activation inhibits inflammatory gene expression and promotes antibacterial gene expression in DSS-induced colitis. Quantitative real time qRT-PCR of (A) interleukin (IL)-1β, IL-6 and macrophage attractant protein-1 (Mcp-1), and (B) inducible NO synthase (iNOs) and cathelicidin (both in the colonic mucosa) and angiogenin 1 (Ang1) (in the ileal mucosa) of wild-type (WT; black bars) and Fxr-null mice (grey bars). Expression was normalised to glyceraldehyde-3-phosphate dehydrogenase, and each bar represents mean±SEM. Student t test, *p<0.05 compared with DSS vehicle-treated WT mice.
Nitric oxide (NO), angiogenin 1 (Ang1) and cathelicidin are known to have microbicidal properties,31–33 and their increased expression may therefore exert positive effects on inflammation status. We show that inducible NO synthase (iNOs) and cathelicidin in the colon and Ang1 expression in the ileum were significantly induced by INT-747 treatment in WT, but not Fxr-null, mice (figure 4B). These data indicate that FXR activation decreases expression of several inflammatory genes and promotes expression of microbicidal genes most probably contributing to the amelioration of DSS-induced colitis in WT mice.
Fxr activation decreases TNBS-dependent intestinal inflammation
We next verified the ameliorating effects of FXR activation on DSS-induced colitis in another well-known murine IBD model.34 TNBS was administered to WT mice twice with a 7-day interval. This protocol resulted in severe colitis in WT C57BL/6 mice. Again, daily administration of INT-747 decreased body weight loss (figure 5A), infiltration of inflammatory cells, degeneration of the epithelial layer and goblet cell loss (figure 5B), as well as intestinal permeability (figure 5C). In concordance with these results, mRNA expression of proinflammatory cytokines Il-1β and Mcp-1 was decreased, while the antimicrobial peptide iNOs was significantly induced in INT-747-treated mice (figure 5D). These results confirm that FXR activation ameliorates intestinal inflammation in a separate, well-established model of experimental colitis.

INT-747-dependent Fxr activation confers protection against TNBS-induced colitis. (A) Body weight variance (%) of wild-type (WT) mice treated with or without TNBS, in the presence or absence of INT-747 (5 mg/kg/day/mouse). Analysis of variance was performed; significant differences (p<0.05) are indicated by different letters. (B) Representative H&E-stained colonic sections, histology and goblet cell loss score; single-headed arrows point to inflammatory infiltrates, and double-headed arrows to epithelial degeneration. Magnification 100×. Each bar represents mean±SEM. Analysis of variance with Bonferroni post hoc test was performed; significant differences (p<0.05) are indicated by different letters. (C) In vivo intestinal permeability measurements on the day of sacrifice of untreated controls (40% EtOH) and TNBS of vehicle- or INT-747-treated mice. Each data point represents mean±SEM. Analysis of variance with Bonferroni post hoc test was performed; significant differences (p<0.05) are indicated by different letters. (D) Quantitative real time qRT-PCR of interleukin (IL)-1β, macrophage attractant protein-1 (Mcp-1) and inducible NO synthase (iNOs) in the mouse colonic mucosa. Expression was normalised to glyceraldehyde-3-phosphate dehydrogenase, and each bar represents mean±SEM. Student t test, *p<0.05; **p<0.01 compared with TNBS vehicle-treated mice.
FXR activation decreases proinflammatory cytokine expression in intestinal cells
We next explored the possible contribution of enterocytes in improving inflammation status in vitro. We used a well-established cellular model for cytokine expression in human enterocytes, HT29 cells.35 36 As in Caco-2 cells, FXR was expressed exclusively in fully differentiated HT29, and GW4064 induced the FXR target gene ileal bile acid binding protein (IBABP), indicating that FXR is functionally active (online supplementary figure 4C,D). As expected, on TNFα treatment, IL-1β mRNA expression was dramatically induced compared with the control. GW4064 treatment alone did not affect IL-1β expression (figure 6A). Strikingly, the TNFα-mediated induction of IL-1β mRNA expression was almost completely abolished on co-treatment with GW4064, indicating that FXR activation counteracts proinflammatory gene expression of IL-1β in intestinal cells. Expression of NF-κB subunits RelA and p50 (figure 6A) was not decreased on TNFα and GW4064 co-treatment. Therefore, in order to investigate if FXR decreases IL-1β mRNA expression by inhibiting the NF-κB transcriptional activity, HEK293 cells were transfected with a κB reporter plasmid alone or in combination with FXR and RXR, and incubated with TNFα, GW4064 or both (figure 6B). In cells transfected with the κB-reporter alone, TNFα increased NF-κB activity. GW4064 had no effect on basal luciferase expression and TNFα-mediated NF-κB activity. In cells co-transfected with FXR, GW4064 almost completely abolished the TNFα-induced κB-responsive luciferase expression. An FXR mutant defective in ligand binding (W469A) did not repress NF-κB activity, indicating that ligand-activated FXR inhibits NF-κB transcriptional activity in vitro.

FXR activation decreases pro-inflammatory cytokine expression in intestinal cells. (A) Quantitative real time (qRT)-PCR analysis of interleukin (IL)-1β, RelA and p50 expression in differentiated HT29 cells incubated with dimethyl sulfoxide (DMSO) (dark grey bars), GW4064 (1 μM, 24 h, white bars) and TNFα (500 U/ml, 6 h, black bars) or both (light grey bars). (B) Reporter assays of NF-κB transcriptional activity. Cells were treated with DMSO (dark grey bars), GW4064 (1 μM, white bars) and/or TNFα (500 U/ml, black and light grey bars, respectively) for 24 h. Each bar represents mean±SD; Student t test, *p<0.05; **p<0.01 compared with TNFα-treated cells. Data are from a representative experiment out of four performed.
FXR activation decreases TNFα production by human immune cells
As TNFα is a clinically validated target in IBD, we have examined the capacity of INT-747 to inhibit TNFα secretion by different human immune cell types: PBMCs, monocytes and dendritic cells. INT-747 inhibited dose-dependent TNFα secretion by PBMCs, whereas no inhibition was induced by cholic acid and only a marginal one by high concentrations of chenodeoxycholic acid (figure 7A). Similar results were obtained with CD14 monocytes (figure 7B). INT-747 also inhibited the GM-CSF/IL-4-induced differentiation of CD14 monocytes into dendritic cells (figure 7C). Dendritic cells differentiated in the presence of INT-747 showed a markedly reduced capacity to secrete TNFα in response to LPS stimulation (figure 7D). Thus, in vitro addition of INT-747 inhibits TNFα secretion by different human and mouse immune cell types, in particular professional antigen-presenting cells.

IINT-747-dependent FXR activation decreases TNFα production by human inflammatory cells. (A) TNFα secretion by lipopolysaccharide (LPS)-stimulated peripheral blood mononuclear cells (PBMCs) incubated with the indicated concentrations of cholic acid (CA), chenodeoxycholic acid (CDCA) or INT-747 (medium ▼; CA ■; CDCA ◆; INT-747 ●). (B) TNFα secretion by LPS-stimulated CD14 monocytes incubated for 24 h with 10 μM CA or CDCA, or with the indicated concentrations of INT-747. Values are expressed as percentage of control LPS-induced response (PBMCs, 1059±91; monocytes, 1737±80 pg/ml) and represent the mean±SEM from at least three experiments. (C) Monocyte-derived dendritic cells generated after 5 days of culture with granulocyte-macrophage colony stimulating factor (GM-CSF) and interleukin (IL)-4 in the presence or absence of 10 μM INT-747 were double-stained with anti-CD14 and anti-CD1a monoclonal antibodies and analysed by flow cytometry. (D) The same cells were stimulated with LPS for 24 h, and TNFα production was determined (GM-CSF/IL4 vehicle, light grey circles; GM-CSF/IL4 INT-747, black circles). Data are from a representative experiment out of three performed.
In line with these results, in vivo treatment with INT-747 also significantly decreased serum TNFα and IFNγ levels after LPS-induced systemic inflammatory response in phagocytic mononuclear cells obtained from mouse peritoneal exudate cells, without affecting their recruitment (online supplementary figure 5). Together these results indicate that in vitro and in vivo FXR activation inhibits inflammatory responses in various isolated human immune cells.
INT-747-induced FXR activation of ex vivo LPMCs from patients with IBD reduces inflammatory cytokine secretion
As shown above, INT-747 is a potent inhibitor of proinflammatory cytokines in vitro in human immune cells. To assess the capacity of this compound to inhibit key components of the inflammatory response in the human intestine, we examined its capacity to inhibit proinflammatory cytokines produced by activation of resident cells from the inflamed target organ, the LPMCs from IBD tissue. Lymphocytes purified from LPMCs were activated with a cocktail of antibodies targeting CD2 and CD28. INT-747 significantly inhibits IFNγ, IL-17 and TNFα production by LPMCs (figure 8A–C). These results indicate that the anti-inflammatory properties of INT-747 observed in PBMCs, monocytes and dendritic cells from healthy volunteers also occur in LPMCs from patients with IBD.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
INT-747 inhibits IFNγ, IL-17 and TNFα production by lymphocyte-enriched LPMCs from IBD patients. LPMCs from four patients with IBD (two Crohn disease and two ulcerative colitis) were stimulated for 24–72 h with αCD2 and αCD28. (A) IFNγ (72 h), (B) IL-17 (24 h) and (C) TNFα (24 h) secretion was determined by ELISA. Values are expressed as percentage of control responses (IFNγ, 1754±1027; IL-17, 2029±1722; TNFα, 341±118 pg/ml). Student t test, *p<0.05; **p<0.01, compared with medium. CDCA, chenodeoxycholic acid.
Discussion
Current medical options for patients with IBD, including glucocorticoids, 5-aminosalicylic acid and anti-TNFα antibody treatment, reduce the proinflammatory response. Although they have all been shown to interfere with NF-κB signalling in vitro, their mechanism of action in IBD is largely elusive and may depend on mechanisms other than NF-κB inhibition. In addition, these treatments are often accompanied by side effects (sepsis, opportunistic infections, tuberculosis (TBC), lymphoma, diabetes, osteoporosis) and significant numbers of treatment failures, highlighting a need for alternative strategies in the treatment of IBD. Here we describe pharmaceutical activation of FXR as a potential new treatment option for IBD.
In this study, we used two independent murine IBD models: DSS- and TNBS-induced colitis. Although there are differences from human IBD, these models share many clinical and pathological features with human IBD with regard to loss of barrier function and inflammatory response.
INT-747, a potent and selective FXR agonist,37 was efficiently taken up in the enterohepatic circulation and strongly activated FXR in the intestine, as evidenced by marked induction of FXR target gene expression. Our findings revealed that INT-747 significantly decreased the severity of DSS- and TNBS-induced colitis in mice, as indicated by decreased body weight loss, colon shortening, and rectal bleeding as well as improvement in colonic histology. The improvement in DSS-induced IBD was not seen in Fxr-null mice, demonstrating that the amelioration of colitis by INT-747 requires FXR. Fxr-null mice do not appear to be more susceptible to DSS colitis than WT mice, and this may be due to the duration of the DSS protocol, the young age of the mice,11 and the micro-environmental conditions. Nevertheless, the basal intestinal permeability of Fxr-null mice is twice as high as that of WT mice (online supplementary figure 3). This is in line with the greater bacterial translocation observed in Fxr-null mice.15 As IBD is thought to result from both dysregulation of the mucosal immune system and compromised intestinal epithelial barrier function in genetically predisposed individuals,2 38 we explored the ability of FXR to counter-regulate intestinal inflammation at different levels.
Enterocytes are known to serve as immuno-effector cells and are capable of secreting cytokines and chemokines to promote inflammation.39 This results in infiltration of macrophages and other immune cells to the site of intestinal inflammation. FXR activation abrogates the expression of the proinflammatory gene, IL-1β, in human enterocyte-like HT29 cells in vitro, and this finding is in line with the capacity of FXR activation to nearly normalise DSS- and TNBS-induced colonic proinflammatory gene expression and to markedly reduce inflammatory infiltrates. In addition, we show that expression of several antibacterial defence genes was significantly induced by INT-747 treatment in WT mice (figure 4B and 5D), suggesting that FXR activation may help to control bacterial overgrowth. The composition of enteric microflora is linked to the initiation and progression of intestinal inflammation, and the intriguing possibility that FXR plays a role in the modification of the intestinal ecology should be the subject of future studies as a putative possibility for both the pathogenesis and treatment of IBD.
In addition, we observed that INT-747 overcomes the increased intestinal permeability induced by DSS and TNBS in WT mice, in concurrence with decreased endothelial ulceration in the colons of these mice. Several cytokines are known to increase permeability in the intestinal epithelial monolayer (eg, TNFα, IFNγ, IL-4 and IL-13)40 41, by modulating tight junction protein expression and localisation. The observed inhibition of cytokine expression by FXR activation may therefore represent a possible mechanism for the preservation of intestinal permeability. Alternatively, FXR may have a direct effect on the integrity of the intestinal mucosa. This hypothesis is supported by in vitro studies showing that FXR activation decreased DSS-induced detachment of human enterocyte-like Caco-2 cells from the monolayer. At this stage, the precise mechanisms by which FXR improves barrier integrity are still unclear.
In addition to aberrant immune response and loss of intestinal barrier integrity, patients with IBD display various degrees of goblet cell loss.8 Goblet cells are intestinal mucin-secreting cells that form the mucus layer that protects the mucosal surface from antigens and thus maintains intestinal barrier function. Also in our experimental colitis models, significant goblet cell loss was detected, which was partially prevented by INT-747, suggesting that FXR activation may also ameliorate intestinal barrier function at the level of goblet cell count.
In agreement with Vavassori et al,11 we show that INT-747-dependent FXR activation inhibits the inflammatory response in immune cells. However, we extend their results obtained in mice and human immortalised immune cell lines to various primary human immune cell types (PBMCs, CD14 monocytes and monocyte-derived dendritic cells). Moreover, we show that the anti-inflammatory properties of FXR activation also reduce inflammatory signalling in LPMCs isolated from patients with IBD. In addition, FXR activation prevented the in vitro differentiation of dendritic cells. These results are in line with the marked reduction in inflammatory infiltrates in our models of murine colitis.
In conclusion, our results indicate that FXR is an important player in the counter-regulation of intestinal inflammation. Although the exact mechanism is still unclear, it is very likely that the protection against experimental colitis may not rely on a single FXR-dependent mechanism. The multi-level protection against intestinal inflammation provides a clear rationale to further explore FXR agonists as a novel therapeutic strategy for IBD. Currently, phase 1 and 2 studies with INT-747 are being performed in patients with metabolic and chronic liver diseases, indicating the safe applicability of this class of compounds for the treatment of human disease.42
Acknowledgments
We thank Lorena Salvatore for expert histology and microscopy assistance. Martin de Smet is acknowledged for his assistance with the biliary bile salt measurements.
References
Supplementary materials
online only appendix
Files in this Data Supplement:
- Fxr is expressed and activated by INT-747 in ileum and colon of WT mice
- Fxr is expressed and activated by INT-747 in ileum and colon of WT mice
- INT-747 ameliorates clinical symptoms and inhibits secretion of inflammatory mediators in DSS-induced murine colitis
- INT-747 ameliorates clinical symptoms and inhibits secretion of inflammatory mediators in DSS-induced murine colitis
- INT-747 ameliorates clinical symptoms and inhibits secretion of inflammatory mediators in DSS-induced murine colitis
- INT-747 ameliorates clinical symptoms and inhibits secretion of inflammatory mediators in DSS-induced murine colitis
- INT-747 ameliorates clinical symptoms and inhibits secretion of inflammatory mediators in DSS-induced murine colitis
- INT-747 ameliorates clinical symptoms and inhibits secretion of inflammatory mediators in DSS-induced murine colitis
- INT-747 ameliorates clinical symptoms and inhibits secretion of inflammatory mediators in DSS-induced murine colitis
- Fxr-null mice have impaired intestinal barrier
- FXR is expressed and functionally active in differentiated enterocyte-like Caco-2 and HT29 cells
- FXR is expressed and functionally active in differentiated enterocyte-like Caco-2 and HT29 cells
- FXR is expressed and functionally active in differentiated enterocyte-like Caco-2 and HT29 cells
- FXR is expressed and functionally active in differentiated enterocyte-like Caco-2 and HT29 cells
- INT-747 inhibits inflammatory responses in vivo
- INT-747 inhibits inflammatory responses in vivo
- INT-747 inhibits inflammatory responses in vivo
Online only appendix
Files in this Data Supplement:
Footnotes
See Commentary, p 432
Linked articles 233304.
Funding LWJK is funded by the University Utrecht High Potentials program, and AM by the Italian Association for Cancer Research (AIRC, Milan, Italy), European Research Council Starting Independent Grant Ideas, Italian Ministry of Health and Education (FIRB- IDEAS 2008 RBID08C9N7), European Community's Seventh Framework Programme FP7/2007– 2013 under Grant Agreement No 202272 (LipidomicNet), Cariplo Fundation Milan, Telethon Fundation (GGP08259), University of Bari. SM is a fellow of CariSPAQ (L'Aquila, Italy). SWCVM is funded by the Netherlands Organisation for Scientific Research (NWO).
Competing interests GP, GL and LA are with Intercept Pharmaceuticals. The other authors declare no competing interests.
Ethics approval This study was conducted with the approval of the Istituto Clinico Humanitas, Milan, Italy.
Provenance and peer review Not commissioned; externally peer reviewed.