Article Text

Abstract
Background The selection of patients for genetic testing to rule out Lynch syndrome is currently based on fulfilment of at least one of the revised Bethesda criteria followed by mismatch repair (MMR) status analysis. A study was undertaken to compare the present approach with universal MMR study-based strategies to detect Lynch syndrome in a large series of patients with colorectal cancer (CRC).
Methods 2093 patients with CRC from the EPICOLON I and II cohorts were included. Immunohistochemistry for MMR proteins and/or microsatellite instability (MSI) analysis was performed in tumour tissue. Germline MLH1 and MSH2 mutation analysis was performed in patients whose tumours showed loss of MLH1 or MSH2 staining, respectively. MSH6 genetic testing was done in patients whose tumours showed lack of MSH6 expression or a combined lack of MSH2 and MSH6 expression but did not have MSH2 mutations. PMS2 genetic testing was performed in patients showing isolated loss of PMS2 expression. In patients with MSI tumours and normal or not available MMR protein expression, all four MMR genes were studied.
Results A total of 180 patients (8.6%) showed loss of expression of some of the MMR proteins and/or MSI. Four hundred and eighty-six patients (23.2%) met some of the revised Bethesda criteria. Of the 14 (0.7%) patients who had a MMR gene mutation, 12 fulfilled at least one of the revised Bethesda criteria and two (14.3%) did not.
Conclusions Routine molecular screening of patients with CRC for Lynch syndrome using immunohistochemistry or MSI has better sensitivity for detecting mutation carriers than the Bethesda guidelines.
- Cancer genetics
- polyposis
- colorectal neoplasia
- colorectal carcinoma
- colorectal cancer genes
- colorectal cancer
- cancer
- cancer prevention
- cancer epidemiology
- colon carcinogenesis
- colonic neoplasms
- cancer susceptibility
- cancer syndromes
- gastrointestinal cancer
- endoscopy
- DNA microsatellite instability
- methylation
Statistics from Altmetric.com
- Cancer genetics
- polyposis
- colorectal neoplasia
- colorectal carcinoma
- colorectal cancer genes
- colorectal cancer
- cancer
- cancer prevention
- cancer epidemiology
- colon carcinogenesis
- colonic neoplasms
- cancer susceptibility
- cancer syndromes
- gastrointestinal cancer
- endoscopy
- DNA microsatellite instability
- methylation
Significance of this study
What is already known about this subject?
Lynch syndrome is one of the most common hereditary cancer syndromes and prevention of colorectal cancer is feasible; however, Lynch syndrome is commonly undiagnosed.
The current strategy for the selection of patients for genetic testing in Lynch syndrome is based on fulfilment of the revised Bethesda criteria followed by mismatch repair system assessment.
There is increasing evidence supporting the role of universal molecular screening in all patients with colorectal cancer in order to improve the detection rate of mutation carriers.
What are the new findings?
In our large population-based cohort with a low prevalence of Lynch syndrome, universal molecular screening shows a better sensitivity for the detection of Lynch syndrome that is not counterbalanced by a significantly lower specificity or accuracy.
Universal molecular screening is able to detect more cases with probably non-sporadic mismatch repair defective colorectal cancer. These cases and their first-degree relatives should undergo the same surveillance and screening schedules as patients with Lynch syndrome.
How might it impact clinical practice in the foreseeable future?
Our findings suggest that, in order to improve the detection of Lynch syndrome and to prevent cancer in mutation carriers, universal molecular screening for MMR deficiency should be routinely performed in all diagnosed colorectal cancers.
Introduction
Lynch syndrome is the most common cause of hereditary colon cancer and is caused by germline mutations in the DNA mismatch repair genes (MMR), mainly MLH1, MSH2 and also MSH6 and PMS2.1 Defects in this pathway lead to changes in the length of nucleotide repeat sequences of tumour DNA, a phenomenon called microsatellite instability (MSI) which constitutes the molecular hallmark of this disease.2 3
Since the molecular characterisation of Lynch syndrome was established, the identification of gene carriers has become of critical importance because of the clinical implications in cancer prevention and surveillance.4 The consensus criteria for the diagnostic strategy of Lynch syndrome is based on selecting out patients who fulfil the Amsterdam criteria or any of the revised Bethesda guidelines5 followed by MSI testing or immunohistochemical (IHC) staining of MMR proteins. Patients whose tumours show MSI or lack of expression of any of these proteins must undergo genetic testing.6 7 However, only 15% of MSI tumours have a hereditary origin and the remaining 85% are sporadic due to epigenetic silencing of MLH1 because of hypermethylation of its promoter region.3 8 Consequently, MLH1 methylation analysis should be performed when MSI is present.9
The main objective of this study was to compare the performance of two different strategies—the current strategy which preselects for testing according to the revised Bethesda guidelines versus universal MMR status study in a large population from two combined cohorts of colorectal cancers (CRC) to select patients for genetic testing to rule out Lynch syndrome.
Methods
Patients
A total of 2093 patients with CRC were included in the study. These patients belong to two separate cohorts of patients included in two nationwide multicentre prospective studies in Spain. The EPICOLON I has been previously described7 10 and included 1222 consecutive patients diagnosed with CRC between November 2000 and October 2001. The EPICOLON II project was a nationwide study aimed at investigating different aspects related to the diagnosis of hereditary CRC. Characteristics of the EPICOLON II cohort have already been described.11 In this study we included 871 consecutive patients with CRC with available tumour tissue from the EPICOLON II cohort diagnosed in 14 Spanish hospitals between March 2006 and December 2007. The study was approved by the institutional ethics committee of each participating hospital. Written informed consent was obtained from all patients.
Immunohistochemistry (IHC)
IHC analysis of the four MMR proteins MLH1, MSH2, MSH6 and PMS2 was performed on two tissue microarrays (TMAs) containing a total of 1895 CRC tumours. Representative tumour regions were identified and marked on H&E-stained slides and subsequently identified in the corresponding tissue blocks. Tissue cylinders of 1 mm in diameter were punched out from the marked areas of each block and incorporated into a recipient paraffin block using the tissue array (Beecher Instruments, Sun Prairie, WI, USA). TMAs contained between 30 and 50 cores of 1 mm needle size. At least two ascertainable cores of tumour tissue were required per case for inclusion in the study. Sections 4 μm thick were cut from TMAs. Slides were placed in a TechMate 500 immunostainer and incubated for 30 min at room temperature with primary antibodies MLH1 (BD Pharmingen, Franklin Lakes, NJ, USA, clone G168-15, dilution 1:30), MSH2 (BD Transduction Laboratories, Franklin Lakes, NJ, USA, clone 44, dilution 1:100), MSH6 (Calbiochem, Darmstadt, Germany, clone FE11, dilution 1:30) and PMS2 (BD Pharmingen, Franklin Lakes, NJ, USA, clone A16-4, dilution 1:100). Antibodies were detected by the Envision+ technique (Dako, Carpinteria, CA, USA). Processed IHC slides were evaluated by two pathologists (CA and AP). A tumour was considered to have normal expression for MLH1, MSH2, MSH6 and PMS2 when unequivocal nuclear staining was seen in some neoplastic epithelial cells, with or without cytoplasmic staining. Cases were classified with loss of expression when there was lack of expression in tumour cells in the presence of internal positive controls (stromal cells or blood vessels). Samples were not scored when no staining of internal control was seen. All cases with lack of correlation between MSI and IHC were re-evaluated and new IHC staining was performed.
Microsatellite instability (MSI)
MSI analysis was performed in 1905 patients. Tumour tissue for MSI analysis was obtained from endoscopic or surgical samples, immediately frozen in liquid nitrogen and stored at −80°C until use. In those cases in which frozen tissue was not available, archival formalin-fixed paraffin-embedded samples were used. Genomic DNA was isolated using the QuiaAmp tissue kit (Quiagen, Cortabeuf, France). MSI status was assessed using BAT26 and NR24 quasimonomorphic markers as previously described.12 13 Tumours were classified as MSI when either of the two markers was unstable.
Methylation specific multiplex ligation-dependent probe analysis
Methylation analysis of the MLH1 gene was performed using methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) according to the manufacturer's protocol using the SALSA MS-MLPA kit ME011 MMR genes (MRC-Holland, Amsterdam, The Netherlands) as previously described.9 The dichotomisation threshold to distinguish methylated versus non-methylated samples was established at 15%.14 15
Germline mutations
The selection of patients and genes for germline mutation analysis was based on IHC and MSI results. Accordingly, MLH1 and MSH2 mutational analysis was performed in all tumours with MLH1 and MSH2 negative staining. MSH6 germline analysis was done in patients with isolated lack of MSH6 expression or combined lack of MSH2 and MSH6 not showing MSH2 mutation. Genetic testing for PMS2 was performed in isolated loss of PMS2 expression tumours. Therefore, germline genetic testing was done often in just one gene. Patients with loss of MSH2 expression with no mutation detected were analysed for EpCAM rearrangements. In patients showing MSI and whose IHC analysis could not be performed, genetic testing of all four MMR genes (MLH1, MSH2, MSH6 and PMS2) was carried out.
Germline mutation studies were performed on genomic DNA isolated from peripheral blood leucocytes or from non-tumour colon tissues. Detection of point mutations was conducted using PCR and direct sequencing of the whole coding sequence and intron-exon boundaries of each gene. Large rearrangements (deletions and insertions) were tested by MLPA (MLPA Kits P003: MLH1-MSH2; P248 (MLH1-MSH2 confirmation), P008 (PMS2-MSH6) and P072 (EpCAM) MRC-HOLLAND; Amsterdam, The Netherlands) following the manufacturer's protocol. The interpretation of genetic analysis results was based on the ACMG Recommendations for Standards for Interpretation of Sequence Variations (2000) and the InSiGHT database.16 Unclassified variants can be seen in table 1 in the online supplement.
Data management and analysis
Analysis was carried out using SPSS software Version 15.0. For continuous variables, relevant measures of central tendency (means for normally distributed data and medians and IQR for skewed data) were used to explore the data. A p value of <0.05 was considered significant. Performance characteristics of screening strategies based on MSI testing and/or protein immunostaining, either directly or through previous selection of patients according to the revised Bethesda guidelines, were calculated with respect to the diagnosis of HNPCC associated with MSH2/MLH1/MSH6/PMS2 germline mutations. Comparisons of paired proportions for sensitivity, specificity and overall accuracy were performed with the McNemar test.
Results
A total of 2093 patients with CRC were included in the study and IHC and/or MSI analysis of MMR proteins were performed. The mean age at diagnosis was 70.5 years (range 26–101); 60% of the patients were male. Thirty-five (1.7%) fulfilled the Amsterdam II criteria and 476 (22.7%) met at least one of the revised Bethesda guidelines. A flow diagram of the analytical strategy and the main results of the study are shown in figure 1.

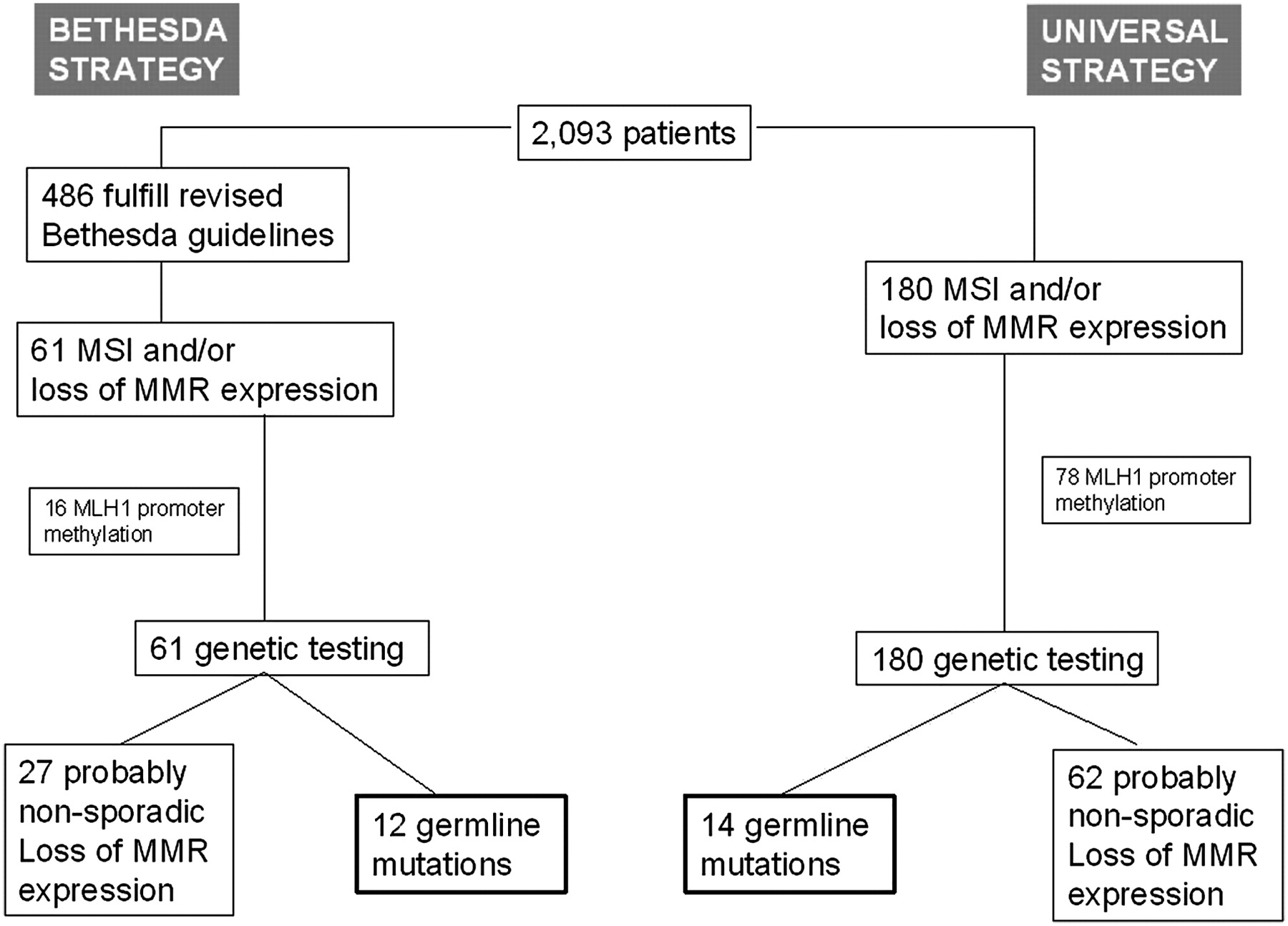{kind=link}
Flow diagram of patients and comparison between Bethesda guidelines and universal molecular screening for the detection of Lynch syndrome mutation carriers. MMR, mismatch repair; MSI, microsatellite instability.
One hundred and eighty patients (8.6%) had loss of MMR protein expression and/or MSI and underwent genetic testing. A total of 155 patients (7.4%) showed loss of expression of any of the MMR proteins. Loss of MLH1 expression was seen in 115 patients (5.5%). MLH1 IHC was considered non-ascertainable in 12 cases (0.6%). In 107 of the MLH1 negative tumours (93.0%), PMS2 immunochemical staining was also negative. Twenty-nine patients (1.4%) had tumours with loss of MSH2 expression, seven of them had isolated loss of MSH2 and 22 had a combined MSH2/MSH6 loss of expression. MSH6 expression was negative in 31 cases (1.6%) and it was not possible to assess MSH6 IHC in 146 cases (7.0%). In nine cases there was an isolated loss of MSH6 expression and the remaining cases showed a combined loss of MSH2/MSH6 expression. Finally, two patients showed an isolated tumoral loss of PMS2 expression. MSI was detected in 152 patients (7.3%), 127 of whom (83.6%) also showed IHC loss of expression of any of the MMR proteins.
Methylation-specific MLPA (MS-MLPA) of MLH1 was performed in cases with tumoral loss of MLH1 expression in order to identify sporadic cases. MLH1 was methylated in 78 cases (67.8%) and MLH1 methylation was non-ascertainable in four cases (4.3%).
Fourteen patients (0.7%) showed a germline pathogenic mutation in one of the MMR genes. Features of the probands are shown in table 1. None of the corresponding families had been previously diagnosed with Lynch syndrome. MSH2 was the most frequently mutated gene (n=7). Four of the patients showed a germline deleterious mutation in MLH1 and three had a mutation in MSH6. None of the patients showed PMS2 or EpCAM pathogenic mutations. None of the germline pathogenic mutations was found in more than one patient. The mean age at diagnosis was 62.8 years (range 28–81) and nine of them were women (64.3%). Of the 14 probands, seven (50%) fulfilled the Amsterdam II criteria and 12 fulfilled the revised Bethesda criteria (85.7%). Only four (28.6%) of the probands were younger than 50 years. None of the four patients with Lynch syndrome and loss of MLH1 expression showed MLH1 promoter methylation.
Characteristics of the probands with germline mutation in a mismatch-repair gene
Patients without a germline mutation in an MMR gene but whose tumours showed loss of MSH2, MSH6, isolated loss of PMS2 and loss of MLH1 without methylation were considered as probable non-sporadic MSI cases. These cases should be sent for genetic counselling and they should receive a similar surveillance schedule to that of confirmed cases of Lynch syndrome with a pathogenic mutation. Moreover, surveillance is also recommended for their first-degree relatives. We detected 62 of these cases; 29 showed loss of MLH1 expression and no methylation, 19 cases had a combined loss of MSH2 and MSH6, four cases had isolated loss of MSH2, eight cases had isolated loss of MSH6 and two cases had isolated loss of PMS2. The clinical characteristics of these patients are shown in tables 2 and 3. The mean age at diagnosis was 67.1 years (range 32–100) and 33 (53.2%) were women. Only five of them (8.1%) fulfilled the Amsterdam II criteria and 27 (41.9%) fulfilled at least one of the revised Bethesda criteria.
Characteristics of patients without germline mutation with tumours showing loss of MLH1 expression and no MLH1 promoter methylation
Characteristics of patients without germline mutation with tumours showing loss of MS2, MSH6 or isolated loss of PMS2
Figure 1 compares the results of the universal IHC strategy with the Bethesda guidelines strategy. According to our data, the revised Bethesda guidelines strategy failed to detect two cases (14.3%) with Lynch syndrome and 36 cases (57.1%) with probable non-sporadic MSI tumours.
Table 4 shows sensitivity, specificity, positive predictive value, negative predictive value and number needed to test for the different strategies to detect germline mutations. It includes the clinical and molecular tools alone or combined.
Value of different strategies for the detection of germline mutations in non-selected CRC population
Discussion
In this study we compare the yield of two strategies in the detection of patients with Lynch syndrome—a universal strategy performing MMR analysis through MSI testing and/or IHC in all cases with CRC versus the strategy of performing tumour tests only when any of the revised Bethesda criteria are identified. Our results show that the universal strategy provides better sensitivity as 14.3% of patients with Lynch syndrome do not fulfil the revised Bethesda guidelines. Moreover, after refinement of the selection of patients for genetic testing performing methylation analysis of MLH1 by MS-MLPA,9 this improvement in sensitivity is not counterbalanced by important differences in specificity or accuracy. With these results we conclude that universal screening for MMR deficiency should be recommended for improving the detection of patients with Lynch syndrome.
Other studies have previously compared similar strategies for CRC screening. A previous study from our group7 showed that the revised Bethesda guidelines were able to detect all cases of Lynch syndrome and we proposed a sequential strategy for first selecting patients at risk with the revised Bethesda guidelines and then refining the selection using either MSI testing or MMR proteins IHC. A limitation of that study was that only MLH1 and MSH2 IHC was performed and therefore only mutations in MLH1 and MSH2 were studied. Other previous studies had included the analysis of MSH6 and PMS2 mutations,17 18 and they showed lack of accuracy of the revised Bethesda guidelines, especially in cases with germline mutation in these two genes. Cost-effectiveness studies also support the recommendation to offer testing for Lynch syndrome to all newly diagnosed patients with CRC.19 For this reason, a panel of experts has recently supported the performance of universal molecular testing to pre-screen for Lynch syndrome using either MSI or MMR proteins IHC.20 Our results support this recommendation, demonstrating the better yield of universal Lynch syndrome screening even in a large population-based cohort with a low prevalence of Lynch syndrome (only 0.7% of CRC cases).
In our cohort we found a relatively low frequency of both MMR-deficient CRC and Lynch syndrome. The proportion of MMR-deficient tumours was 8.6%, lower than the 10–15% commonly described. Dietary or ethnic factors may account for this difference. Studies done in the Mediterranean area have shown lower rates of MSI than others in North America or Northern Europe.21 22 It is thought that dietary, toxic or other environmental factors can cause epigenetic disruption of MLH1 such as promoter hypermethylation of the gene, influencing in this manner the proportion of MMR-defective tumours.6 23 This could explain the lower rates of MSI cases in the Mediterranean countries where some dietary habits that could be related to MSI—such as red meat ingestion, cooking practices that increase the intake of heterocyclic amines (like frying, barbecue or broiling) or consumption of high-grade alcoholic drinks—are less frequent.24 25 On the other hand, ethnic factors may also have some influence. A recent study found that 9.9% of tumours from Hispanics living in the USA were MSI,26 a similar proportion to the tumours reported in our series. However, another study from our group found a high proportion of MSI tumours in a Peruvian group of patients.27 Together, this suggests the high variability in the proportion of patients with MMR-deficient CRC as multiple factors can influence this proportion. Moreover, the proportion of patients with Lynch syndrome is also lower than that described in previous studies. There are few population-based studies on the prevalence of Lynch syndrome, which ranges between 2% and 2.8%.17 18 28 29 Differences in the prevalence of genetic diseases can be explained by differences in genetic background among populations, especially in syndromes where the penetrance is incomplete and other genetic and environmental factors may act as penetrance modifiers.30 Our prevalence is among the lowest described so far, but there are some differences in our population that could explain this, at least in part. One important difference is the geographical extension of the studies, some of which referred to small areas (ie, nine small hospitals from south-east Finland28 29 and six hospitals from the Columbus, Ohio metropolitan area),17 18 whereas our study is a nationwide multicentre study involving 20 and 14 hospitals all over Spain in the EPICOLON I and II cohorts, respectively. A possible consequence of these different study populations is a higher frequency of recurrent mutations in previous studies that might underlie founder effects. Specifically, 50% of probands (14/28) in the Finnish studies share the same MLH1 mutation and only three MLH1 mutations account for 82% of Lynch syndrome cases.28 29 In the Ohio studies17 18 the frequency of repeated mutations is lower, but still 24% of probands (10/41) have the same MSH2 mutation and almost one-third (13/41) of the Lynch syndrome cases are accounted for by only two MSH2 mutations. On the contrary, in our cohort there are no repeated mutations. This is probably due to the wider geographical area from which patients were recruited in our study, which presumably can result in a more varied origin and genetic background. On the other hand, although founder mutations have been described for MLH1 and MSH2 genes in the Spanish population,31–33 none of these mutations has been found in the present series. In some of the cases a germline mutation should be expected because of the existence of loss of MSH2 and/or MSH6 expression or loss of MLH1 without promoter methylation (tables 2 and 3). In any event, a false positive result for IHC, especially when accompanied by normal MSI, would help account for cases in which no mutation was found.
There are several reasons for performing molecular pre-screening for detecting Lynch syndrome. Lynch syndrome is one of the most common autosomal dominant cancer conditions and appropriate screening and surveillance for CRC and other cancers can prevent incidence and mortality.4 However, Lynch syndrome is underdiagnosed,34 mainly because of the lack of awareness of the Bethesda guidelines. Fulfilment of the Amsterdam criteria or revised Bethesda guidelines requires a detailed family history and a number of studies have reported that family history assessment is often inadequate in clinical practice, even in specialised cancer centres.35–37 Thus, the number of patients who undergo genetic testing for Lynch syndrome is lower than expected. This could be due to patients' lack of knowledge about their family history, small family size, frequent lack of documentation of cancer cases and a remarkable lack of good family history-taking by clinicians.38 39 Moreover, the recognition of the Bethesda criteria requires a very high level of cooperation between pathologists and clinicians and, to date—more than 10 years after the description of the Bethesda guidelines40—their utilisation in clinical practice is very scarce. Finally, there are clinical41–43 and pathological44 predictive models for the identification of persons at risk for Lynch syndrome, but its use is also low. Clinical web-based predictive models such as PREMM1,2,6 have demonstrated its accuracy in the selection of patients for genetic testing and have been validated in population-based studies.45 On the other hand, the performance of universal Lynch syndrome pre-screening with MSI or MMR protein IHC has additional benefits. Apart from screening for Lynch syndrome, testing for MSI is important because of its prognostic and possible therapeutic implications. According to some population-based studies, high-frequency MSI is associated with prolonged survival independently of classic clinical prognosis factors including disease stage. The considerable survival advantage conferred by high-frequency MSI appears to be applicable to both inherited and sporadic types of CRC.46 In addition, the obtained benefit achieved by fluorouracil-based adjuvant chemotherapy in patients with TNM stages II and III seems to be limited to those with stable tumours but not to patients whose tumours are MSI.47–51 In this sense, recent evidence shows that the lack of efficacy of 5-fluorouracil is related to the CIMP phenotype more than the MSI status.52 Knowledge of the MSI status could therefore also help physicians to assess the prognosis and guide treatment choices.
Another advantage of universal screening is the detection of CRC cases with probable non-sporadic loss of expression of MMR proteins. Cases with loss of MSH2, MSH6 or isolated loss of PMS2 are usually caused by germline mutations in one of these genes and, at this time, no other form of sporadic inactivation of these genes has been described. In these cases, when germline mutation in a MMR gene is not found, the cause of MMR deficiency is unknown but probably it is not sporadic. Another group of cancers where the cause of MMR gene silencing is unclear is when there is loss of MLH1 expression without methylation of MLH1 promoter and without MLH1 germline mutation. In all these patients, in the absence of an explanation for a sporadic cause for the MMR silencing, a germline mechanism of inactivation should be suspected. These patients are considered to have probable Lynch syndrome and are usually followed up as Lynch syndrome patients and cancer screening is also recommended to their first-degree relatives.53 A subset of these patients with loss of MSH2 expression has recently been explained by germline deletions in the epithelial cell adhesion molecule gene (EpCAM), also known as TACSTD1.54–56 These cases are MSH2-deficient although no MMR gene mutations can be identified, demonstrating the probable hereditary origin and the importance of these probably non-sporadic losses of MMR immunohistochemical expression. In our study, using the Bethesda guidelines strategy, more than half of the cases with probable non-sporadic MMR deficiency would be undetected with the subsequent lack of opportunity for cancer prevention.
In conclusion, routine molecular screening of patients with CRC for Lynch syndrome using MSI analysis or IHC is able to detect more probands with Lynch syndrome than previous selection with the Bethesda guidelines. Moreover, more patients with suspected Lynch syndrome because of a probable non-sporadic loss of MMR protein expression are also detected, providing more opportunities for early detection and prevention of cancer in these patients and their relatives.
Appendix Members of the EPICOLON Consortium (Gastrointestinal Oncology Group of the Spanish Gastroenterological Association)
Hospital 12 de Octubre, Madrid: Juan Diego Morillas (local coordinator), Raquel Muñoz, Marisa Manzano, Francisco Colina, Jose Díaz, Carolina Ibarrola, Guadalupe López, Alberto Ibáñez; Hospital Clínic, Barcelona: Antoni Castells (local coordinator), Virgínia Piñol, Sergi Castellví-Bel, Francesc Balaguer, Victoria Gonzalo, Teresa Ocaña, María Dolores Giráldez, Maria Pellisé, Anna Serradesanferm, Leticia Moreira, Miriam Cuatrecasas, Josep M. Piqué; Hospital Clínico Universitario, Zaragoza: Ángel Lanas (local coordinator), Javier Alcedo, Javier Ortego; Hospital Cristal-Piñor, Complexo Hospitalario de Ourense: Joaquin Cubiella (local coordinator), Mª Soledad Díez, Mercedes Salgado, Eloy Sánchez, Mariano Vega; Hospital del Mar, Barcelona: Montserrat Andreu (local coordinator), Anna Abuli, Xavier Bessa, Mar Iglesias, Agustín Seoane, Felipe Bory, Gemma Navarro, Beatriz Bellosillo; Josep Mª Dedeu, Cristina Álvarez, Begoña Gonzalez; Hospital San Eloy, Baracaldo and Hospital Donostia, CIBERehd, University of Country Basque, San Sebastián: Luis Bujanda (local coordinator) Ángel Cosme, Inés Gil, Mikel Larzabal, Carlos Placer, María del Mar Ramírez, Elisabeth Hijona, Jose M. Enríquez-Navascués y Jose L. Elosegui; Hospital General Universitario de Alicante: Artemio payá (EPICOLON I local coordinator), Rodrigo Jover (EPICOLON II local coordinator), Cristina Alenda, Laura Sempere, Nuria Acame, Estefanía Rojas, Lucía Pérez-Carbonell; Hospital General de Granollers: Joaquim Rigau (local coordinator), Ángel Serrano, Anna Giménez; Hospital General de Vic: Joan Saló (local coordinator), Eduard Batiste-Alentorn, Josefina Autonell, Ramon Barniol; Hospital General Universitario de Guadalajara and Fundación para la Formación e Investigación Sanitarias Murcia: Ana María García (local coordinator), Fernando Carballo, Antonio Bienvenido, Eduardo Sanz, Fernando González, Jaime Sánchez, Akiko Ono; Hospital General Universitario de Valencia: Mercedes Latorre (local coordinator), Enrique Medina, Jaime Cuquerella, Pilar Canelles, Miguel Martorell, José Ángel García, Francisco Quiles, Elisa Orti; CHUVI-Hospital Meixoeiro, Vigo: EPICOLON I: Juan Clofent (local coordinator), Jaime Seoane, Antoni Tardío, Eugenia Sanchez. EPICOLON II Mª Luisa de Castro (local coordinator), Antoni Tardío, Juan Clofent, Vicent Hernández; Hospital Universitari Germans Trias i Pujol, Badalona and Section of Digestive Diseases and Nutrition, University of Illinois at Chicago, IL, USA.: Xavier Llor (local coordinator), Rosa M. Xicola, Marta Piñol, Mercè Rosinach, Anna Roca, Elisenda Pons, José M. Hernández, Miquel A. Gassull; Hospital Universitari Mútua de Terrassa: Fernando Fernández-Bañares (local coordinator), Josep M. Viver, Antonio Salas, Jorge Espinós, Montserrat Forné, Maria Esteve; Hospital Universitari Arnau de Vilanova, Lleida: Josep M. Reñé (local coordinator), Carmen Piñol, Juan Buenestado, Joan Viñas; Hospital Universitario de Canarias: Enrique Quintero (local coordinator), David Nicolás, Adolfo Parra, Antonio Martín; Hospital Universitario La Fe, Valencia: Lidia Argüello (local coordinator), Vicente Pons, Virginia Pertejo, Teresa Sala; Hospital Sant Pau, Barcelona: Dolors Gonzalez (local coordinator) Eva Roman, Teresa Ramon, Maria Poca, Mª Mar Concepción, Marta Martin, Lourdes Pétriz; Hospital Xeral Cies, Vigo: Daniel Martinez (local coordinator); Fundacion Publica Galega de Medicina Xenomica (FPGMX), CIBERER, Genomic Medicine Group-University of Santiago de Compostela, Santiago de Compostela, Galicia, Spain: Ángel Carracedo (local coordinator), Clara Ruiz-Ponte, Ceres Fernández-Rozadilla, Mª Magdalena Castro; Hospital Universitario Central de Asturias: Sabino Riestra (local coordinator), Luis Rodrigo; Hospital de Galdácano, Vizcaya: Javier Fernández (local coordinator), Jose Luis Cabriada; Fundación Hospital de Calahorra (La Rioja) La Rioja: Luis Carreño (local coordinator), Susana Oquiñena, Federico Bolado; Hospital Royo Villanova, Zaragoza: Elena Peña (local coordinator), José Manuel Blas, Gloria Ceña, Juan José Sebastián; Hospital Universitario Reina Sofía, Córdoba: Antonio Naranjo (local coordinator).
References
Footnotes
Funding The present work was supported by grants from Instituto de Salud Carlos III (INT09/208 and PI08/0726) to RJ, from Fundación de la CV para la Investigación en el Hospital General Universitario de Alicante to RJ, LPC, AP and CA, grant SAF 07-64873 from the Ministerio de Educación y Ciencia to AC, grants from the Asociación Española contra el Cáncer (Fundación Científica and Junta de Barcelona) to AC, funds from the AGAUR (2009 SGR 849) to AC. LP-C is a recipient of a predoctoral grant from Instituto de Salud Carlos III (FI07/00303). CIBERehd is funded by the Instituto de Salud Carlos III. CG is a recipient of a predoctoral grant from Conselleria d'Educació de la Generalitat Valenciana (VALi+d). SC-B is supported by a contract (CP 03-0070) from Instituto de Salud Carlos III.
Competing interests NA is a research nurse granted by MSD. None of the other authors have any potential conflicts to disclose.
Ethics approval Ethics approval was provided by CEIC HGU Alicante.
Provenance and peer review Not commissioned; externally peer reviewed.