Article Text

Abstract
Objective Using genome-wide promoter methylation assay, B cell CLL/lymphoma 6 member B (BCL6B) was found to be preferentially methylated in cancer. A study was undertaken to examine the epigenetic regulation, biological function and clinical significance of BCL6B in gastric cancer (GC).
Methods BCL6B promoter methylation was evaluated by combined bisulfite restriction analysis and sequencing. The biological functions of BCL6B were determined by cell viability, colony formation, flow cytometry and in vivo tumorigenicity assays. The molecular targets of BCL6B were identified by cDNA expression array.
Results BCL6B was silenced or downregulated in all nine GC cell lines and readily expressed in normal gastric tissues. Loss of BCL6B expression was regulated by promoter hypermethylation. Re-expression of BCL6B in GC cell lines inhibited colony formation, suppressed cell viability, induced apoptosis and restrained the tumorigenecity in nude mice. These effects were associated with upregulation of the pro-apoptosis genes tumour necrosis factor receptor superfamily member 1A, caspase-8, caspase-9, caspase-3 and caspase-7 and nuclear enzyme poly (ADP-ribose) polymerase, downregulation of the pro-proliferation genes S100 calcium binding protein A4 and vascular endothelial growth factor A, and induction of the tumour suppressor genes ataxia telangiectasia mutated homologue and p53. BCL6B hypermethylation was detected in 49.0% (102/208) and 66.3% (67/101) of two independent cohorts of patients with GC, respectively. BCL6B methylation was an independent factor for the survival of patients with GC (p=0.001 for cohort I, p=0.02 for cohort II).
Conclusions BCL6B plays a pivotal role as a potential tumour suppressor in GC. Detection of methylated BCL6B may serve as an independent biomarker for the prognosis of GC.
- B cell CLL/lymphoma 6 member B
- gastric cancer
- epigenetic alteration
- methylation
- tumour suppressor gene
- prognosis
- gastroscopy
- tumour markers
- methylation
- gene regulation
- molecular carcinogenesis
- molecular oncology
- carcinogenesis
- cancer susceptibility
- cancer immunobiology
- cancer genetics
- cancer prevention
- colorectal adenomas
- gastrointestinal bleeding
- colorectal cancer screening
- cancer
- non-alcoholic steatohepatitis
Statistics from Altmetric.com
- B cell CLL/lymphoma 6 member B
- gastric cancer
- epigenetic alteration
- methylation
- tumour suppressor gene
- prognosis
- gastroscopy
- tumour markers
- methylation
- gene regulation
- molecular carcinogenesis
- molecular oncology
- carcinogenesis
- cancer susceptibility
- cancer immunobiology
- cancer genetics
- cancer prevention
- colorectal adenomas
- gastrointestinal bleeding
- colorectal cancer screening
- cancer
- non-alcoholic steatohepatitis
Significance of this study
What is already known about this subject?
Epigenetic alterations, particularly inactivation of tumour suppressor genes through promoter hypermethylation, play an important role in the initiation and progression of gastric cancer.
BCL6B gene, located on chromosome 17p13.1, is a homologue of B cell CLL/lymphoma 6 and plays a role in the nucleus as a sequence-specific transcriptional repressor.
Expression of human BCL6B mRNA is ubiquitously detected in human tissues.
What are the new findings?
BCL6B is commonly silenced or downregulated in gastric cancer by promoter methylation.
BCL6B acts as a novel tumour suppressor in gastric cancer.
The anti-tumorigenic function of BCL6B was modulated by inducing both intrinsic and extrinsic apoptosis pathways, upregulating tumour suppressors ATM and p53 and suppressing cell proliferation by upregulation of p21 and downregulation of CDK4 and PLAUR.
BCL6B methylation serves as a potential epigenetic biomarker to predict the outcome of patients with gastric cancer.
How might it impact on clinical practice in the foreseeable future?
Detection of methylated BCL6B may serve as a new biomarker for the prognosis of patients with gastric cancer.
Introduction
Gastric cancer (GC) is one of the most common malignancies and remains the second leading cause of cancer-related death worldwide.1 However, the mechanism leading to the development of GC remains unclear. Emerging evidence indicates that epigenetic alterations, particularly inactivation of tumour suppressor genes or tumour-related genes through promoter hypermethylation, play an important role in the development and progression of GC.2–8 The identification of such novel genes targeted by promoter methylation may help to find alternative approaches for diagnostic and therapeutic evaluation. We recently identified a novel preferentially methylated gene, B cell CLL/lymphoma 6 member B (BCL6B), in human cancer through methylation-sensitive representational difference analysis (MS-RDA).9
BCL6B, also known as BAZF, ZNF62 and ZBTB28, is a homologue of B cell CLL/lymphoma 6 (BCL6) and contains the BTB/POZ domain in the NH2 terminal region and zinc finger motifs in the COOH terminal region and the middle portion. It plays a role in the nucleus as a sequence-specific transcriptional repressor.10 This gene is located on chromosome 17p13.1 and is about 800 kb telomeric to the tumour suppressor p53 gene.11 Expression of human BCL6B mRNA is ubiquitously detected in human tissues with abundant expression in the heart and placenta.11 BCL6B is required for secondary responses of memory CD8+ T cells.12 Overexpression of BCL6B induces apoptosis in NIH3T3 cells.13 The role of BCL6B in tumour development is largely unknown. Using MS-RDA,9 we have shown that BCL6B is preferentially methylated in cancer. In this study the epigenetic regulation, biological function, molecular basis and clinic application of BCL6B in GC were examined.
Materials and methods
GC cell lines
Ten GC cell lines (AGS, BGC823, Kato III, MGC803, MKN28, MKN45, NCI87, SNU1, SNU16 and SNU719) were used in this study.7 Cell lines were maintained in RPMI-1640 medium (Gibco BRL, Rockville, Maryland, USA) with 10% fetal bovine serum (Gibco BRL).
Subjects and sample collection
Two cohorts of patients with histologically-confirmed GC were included in the study. The first cohort (cohort I) included 208 patients diagnosed in the First Affiliated Hospital of Sun Yat-sen University in Guangzhou, China and the second cohort (cohort II) included 101 patients diagnosed in the Prince of Wales Hospital at the Chinese University of Hong Kong, Hong Kong. The demographic and clinicopathological features of the patients are shown in table 1. In addition, 12 paired biopsy specimens from primary gastric tumour and adjacent non-tumour sites from patients with GC and 20 biopsy specimens of normal gastric mucosa from healthy controls during endoscopy were obtained at the Prince of Wales Hospital. All subjects provided informed consent for obtaining the study specimens. The study protocol was approved by the Clinical Research Ethics Committee of the Sun Yat-sen University of Medical Sciences and the Ethics Committee of the Chinese University of Hong Kong. Human normal tissue RNA was purchased commercially (Stratagene, La Jolla, California, USA).
Clinicopathological features of BCL6B methylation in 309 patients with gastric cancer in cohorts I and II
Methylation-sensitive representational difference analysis (MS-RDA)
MS-RDA was performed to identify hypermethylated genes in cancer.9 14 Briefly, the genomic DNAs of untreated cancer cell line CNE-1 and CNE-1 treated with 5-Aza-2′-deoxycytidine (5-Aza; Sigma, St Louis, Missouri, USA) (50 Amol/l) were prepared for two cycles of competitive hybridisation. PCR products from the second hybridisation were cloned, sequenced and analysed using the BLASTN algorithm (National Center for Biotechnology Information). A 347 bp fragment located in the position 147926-148272 of AC040977 (NCBI accession number) spanning part of the exon 1 and intron 1 of the BCL6B gene was identified (see figure 1 in online supplement).
RNA extraction, semi-quantitative RT-PCR and real-time PCR analyses
Total RNA was extracted from cell pellets or tissues and cDNA was synthesised (Roche, Indianapolis, Indiana, USA). Semi-quantitative RT-PCR and real-time PCR was performed.15 Primer sequences are listed in table 1 in the online supplement.
Bisulfite treatment of DNA, methylation-specific PCR (MSP) and combined bisulfite restriction analysis (COBRA)
Genomic DNA was extracted and DNA was chemically modified with sodium metabisulphite.15 The bisulfite-modified DNA was amplified by methylation-specific PCR (MSP) using the primer pairs shown in table 1 in the online supplement.15 Combined bisulfite restriction analysis (COBRA) was performed to semi-quantitate the methylated and unmethylated DNA after sodium bisulfite modification.7
Bisulfite genomic sequencing (BGS)
The PCR products of bisulfite-treated DNA were cloned into the pCR4-Topo vector (Invitrogen, Carlsbad, California, USA). Seven to eight colonies were randomly chosen and sequenced. Sequencing analysis was performed by SeqScape software (Applied Biosystems, Foster City, California, USA) and nine CpG sites spanning the −95 to +95 bp region were evaluated (figure 1).

(A) BCL6B was readily expressed in normal human tissues and fetal tissues by semi-quantitative RT-PCR. (B) BCL6B was silenced or downregulated in all gastric cancer (GC) cell lines by RT-PCR. Methylation of BCL6B was determined by methylation-specific PCR (MSP) and combined bisulfite restriction analysis (COBRA). The undigested fragment (upper band) represents the unmethylated DNA (U). The digested fragments represent the methylated DNA (M). (C) A typical CpG island spans the promoter region of BCL6B. Each vertical bar represents a single CpG site. The transcription start site (TSS) is indicated by a curved arrow. A region for combined bisulfite restriction analysis (COBRA) and bisulfite genomic sequencing (BGS) is shown. The BstUI digestive site is indicated. Bisulfite genomic sequencing (BGS) analysis confirmed the methylation status of BCL6B in GC cell lines. (D) BCL6B mRNA expression was restored after treatment with the demethylation reagent 5-aza-2′-deoxycytidine (5-Aza) alone (D1) or in combination with the histone deacetylase inhibitor trichostatin A (TSA) (D2).
5-Aza and trichostatin A (TSA) treatment
GC cells were seeded at a density of 1×106 cells/ml in 10 cm dishes. After overnight culture, cells were treated with 2 μM of the DNA demethylating agent 5-Aza for 96 h. Some cell lines were further treated with the histone deacetylase inhibitor trichostatin A (TSA; 300 nmol/l) for an additional 24 h. After treatment the cells were harvested for DNA and RNA extractions.
Immunohistochemistry
Immunohistochemistry was performed on paraffin sections of GCs using anti-BCL6B antibody (1:400, Abcam, Cambridge, Massachusetts, USA). The extent of BCL6B staining was scored by assigning the percentage of positive tumour cells (0, none; 1, <20% of positive staining cells; 2, 20–50% of positive staining cells; 3, >50% of positive staining cells).
Construction of BCL6B expression plasmid
The full-length open reading frame sequence of BCL6B was obtained by RT-PCR amplification of normal human stomach cDNA. The PCR aliquots were subcloned into the mammalian expression vector pcDNA3.1 containing an HA-tag (Invitrogen) and then verified by DNA sequencing.
Cell viability assay
Cell viability was determined by the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay (Promega, Madison, Wisconsin, USA).
Colony formation assay
Cells were transfected with pcDNA3.1-HA-BCL6B or the empty vector pcDNA3.1 using lipofectamine 2000 (Invitrogen). After 48 h of transfection, cells were selected with G418 at 0.5 mg/ml (Calbiochem, Darmstadt, Germany) for 10–14 days. Colonies (≥50 cells/colony) were counted after staining with 5% crystal violet.
Annexin V apoptosis assay
Apoptosis was assessed by flow cytometry after staining with Annexin V (FITC-conjugated) (BD Biosciences, Erembodegem, Belgium) and 7-amino-actinomycin (7-AAD; BD Biosciences).
In vivo tumorigenicity
BGC823 cells (1×107 cells in 0.1 ml phosphate-buffered saline) transfected with BCL6B or pcDNA3.1 were injected subcutaneously into the dorsal left flank of 4-week-old male Balb/c nude mice (n=5 per group). Tumour diameter was measured every 2 days for 3 weeks. Tumour volume (mm3) was estimated by measuring the longest and shortest diameter of the tumour and calculating as previously described.7 All experimental procedures were approved by the Animal Ethics Committee of the Chinese University of Hong Kong.
cDNA expression array
Gene expression profiles of AGS cells stably transfected with pcDNA3.1-HA-BCL6B or pcDNA3.1 vector were analysed by Human Cancer Pathway RT2 Profiler PCR Array (SABiosciences, Frederick, Maryland, USA). This array contains 84 functionally well-characterised genes involved in human tumorigenesis (http://www.sabiosciences.com). Gene expression with fold changes ≥1.5 or ≤−1.5 was considered to be of biological significance.
Western blot analysis
Total protein was extracted and measured by the DC protein assay method of Bradford (Bio-Rad, Hercules, California, USA). Thirty micrograms of protein from each sample were separated on 12% SDS-PAGE and transferred onto nitrocellulose membranes (GE Healthcare, Piscataway, New Jersey, USA). Blots were immunostained with primary antibody and secondary antibody, respectively.
Statistical analysis
The results are expressed as mean±SD. The Mann–Whitney U test was performed to compare the difference of BCL6B protein expression between tumour and adjacent non-tumour tissues. The difference in tumour growth rate between the two groups of nude mice was determined by repeated-measures analysis of variance. The χ2 test was used for comparison of patient characteristics and distributions of methylation and covariates by vital status. Patient age (at entry to follow-up) by vital status was compared using the t test. Crude RRs of death associated with BCL6B methylation and other predictor variables were first estimated using the univariate Cox proportional hazards regression model. A multivariate Cox model was constructed to estimate the adjusted RR for BCL6B methylation. Overall survival in relation to methylation status was evaluated by the Kaplan–Meier survival curve and the log-rank test. Analysis of overall survival was limited to a 5-year period to avoid the probability of death not related to GC. A p value <0.05 was considered statistically significant.
Results
BCL6B downregulated or silenced by promoter methylation in GC cell lines
As shown in figure 1A, BCL6B transcription was reduced or silenced in all nine GC cell lines examined (100%) but was readily expressed in the normal human tissues including stomach tissue and fetal tissues, inferring aberrant gene silencing of BCL6B in GC.
It has been well documented that aberrant promoter CpG methylation is related to gene silencing. We next explored BCL6B methylation status by MSP and COBRA (figure 1B). Full or partial methylation was detected in eight GC cell lines (AGS, BGC823, Kato III, MGC803, MKN28, MKN45, SNU1 and SNU719) which showed silenced or downregulated BCL6B expression while methylation was not detected in SNU16 with reduced BCL6B expression (figure 1B). A good correlation between downregulation/silencing and methylation of BCL6B was observed in these cell lines. The detailed methylation status of individual CpG sites was examined by high-resolution BGS. The BGS results confirmed those of the MSP and COBRA analyses (figure 1C).
Pharmacological demethylation restores BCL6B expression
GC cell lines (MKN28, NCI86, AGS and SNU719) were treated with the DNA methyltransferase inhibitor 5-Aza. Re-expression of BCL6B was observed in all cell lines examined (figure 1D). As treatment of 5-Aza only leads to partial reactivation of BCL6B in SNU719 and NCI87 cells, these two cell lines were further treated with TSA. More restored expression of BCL6B was observed in SNU719 and NCI87 treated with 5-Aza together with TSA than with 5-Aza only (figure 1D2).
Downregulation of BCL6B protein in primary GCs
Immunohistochemistry was performed to evaluate BCL6B protein expression in 12 paired gastric tumours and their adjacent non-tumour tissues (figure 2A). BCL6B protein was expressed predominantly in the nucleus and also detectable in the cytoplasm in adjacent non-tumour tissues but was significantly downregulated in primary tumour tissues (p<0.001, figure 2B).

(A) Representative images of BCL6B protein expression in gastric cancer and their adjacent non-tumour tissues determined by immunohistochemistry (IHC). (B) BCL6B protein was downregulated significantly in primary tumour specimens compared with the adjacent non-tumour tissues by IHC (p<0.001, n=12). IHC scoring was performed according to the percentage of positive tumour cells (1, <20%; 2, 20–50%; 3, >50%).
Ectopic expression of BCL6B suppressed GC cell growth
The frequent silencing of BCL6B in GC cell lines and primary cancers but not in normal gastric mucosa suggests that BCL6B is probably a tumour suppressor. In this connection we examined the effect of ectopic expression of BCL6B on the growth and viability in AGS and BGC823 cells with complete methylation and silencing of BCL6B. Re-expression of BCL6B in the stably transfected AGS and BGC823 cells was confirmed by RT-PCR (figure 3A1) and western blot analysis (figure 3A2). Ectopic expression of BCL6B caused a significant decrease in cell viability in AGS (p<0.001) and BGC823 (p<0.001, figure 3B). The inhibitory effect on GC cell growth was further confirmed by colony formation assay. The colonies formed by BCL6B-transfected cells were significantly fewer and smaller than those formed by empty vector-transfected cells, with 56% reduction in AGS (p<0.001) and 78% reduction in BGC823 (p<0.001, figure 3C). Thus, BCL6B exhibits growth inhibitory ability in GC cells and functions as a potential tumour suppressor.

Effect of ectopic BCL6B expression on tumour growth. (A) Ectopic expression of BCL6B mRNA (A1) and protein (A2) in AGS and BGC823 cell lines was evidenced by RT-PCR and western blot analysis, respectively. (B) BCL6B significantly inhibited cell viability in AGS (p<0.001) and BGC823 cells (p<0.001). (C) The effect of BCL6B on cancer cell growth was further confirmed by colony formation assay. The left panel shows the representative images of the colony formation in gastric cancer cells transfected with pcDNA3.1/BCL6B or empty vector (pcDNA3.1). Quantitative analysis of colony numbers is shown in the right panel. Data are mean±SD; **p<0.001. (D) BCL6B inhibited growth of tumours derived from BGC823 in vivo. (D1) Tumour growth curve of BCL6B-expressing cells in nude mice compared with vector (pcDNA3.1)-transfected cells. Data are mean±SD (n=5/group) of three separate experiments. (D2) Histogram showing the mean tumour weights of the BCL6B and vector groups. *p<0.05.
BCL6B inhibits tumour growth in nude mice
To confirm the tumour suppressive effect of BCL6B in GC, we tested whether BCL6B could suppress the growth of GC cells in nude mice in vivo. The tumour growth curve of BGC823 stably transfected with BCL6B or empty vector in nude mice is shown in figure 3D. The tumour volume was significantly smaller in BCL6B-transfected nude mice than in vector control mice (p<0.0001, figure 3D1). At the end of the experiment xenograft tumour was isolated. The mean tumour weight was significantly less in BCL6B-transfected nude mice than in vector control mice (p<0.01, figure 3D2), indicating that BCL6B acts as a tumour suppressor in GC.
BCL6B-induced cell apoptosis
Suppression of cell growth in tumour cells is usually associated with concomitant activation of cell death pathways. We therefore examined the contribution of apoptosis to the observed growth inhibition of BCL6B-transfected cells using flow cytometry with Annexin V and 7-AAD double staining (figure 4A1). Our results showed an increase in the numbers of both early apoptotic cells (13.14±0.65% vs 6.58±0.52%, p<0.001) and late apoptotic cells (5.82±0.63% vs 4.30±0.16%, p<0.05) in BCL6B-transfected AGS cells compared with AGS cells transfected with control vector (figure 4A2). This effect was also observed in BCL6B-transfected BGC823 cells, the proportions of both early apoptotic cells (15.99±0.15% vs 10.42±0.63%, p<0.0001) and late apoptotic cells (6.86±0.42% vs 4.53±0.62%, p<0.01) being significantly increased compared with the control vector-transfected BGC823 cells (figure 4A2).

BCL6B induced apoptosis in gastric cancer cell lines. (A) BCL6B induced apoptosis in AGS and BGC823 cells by flow cytometry analysis following Annexin V and 7-AAD staining. (A1) Representative FACS images of gastric cancer cells transfected with pcDNA3.1 or BCL6B. (A2) Quantitative analyses of early apoptotic and late apoptotic cells. The experiment was repeated three times in triplicate. Data are mean±SD, *p<0.05; **p<0.001, ***p<0.0001. (B) Effect of BCL6B on protein expression of the active form of pro-apoptosis regulators in AGS and BGC823 cells examined by western blot analysis. (C) Effect of BCL6B on protein expression of the tumour suppressor genes ataxia telangiectasia mutated homologue (ATM) and p53 by western blot analysis.
To elucidate the molecular basis of apoptosis we examined the pro-apoptosis mediators, including the active form of caspase-3, caspase-7, caspase-8, caspase-9 and nuclear enzyme poly (ADP-ribose) polymerase (PARP) in the stably transfected AGS and BGC823 cells. Ectopic expression of BCL6B significantly increased the protein levels of the active form of caspase-3, caspase-7, caspase-8, caspase-9 and PARP, suggesting that BCL6B induced apoptosis through both intrinsic and extrinsic caspase-dependent pathways (figure 4B).
Identification of genes modulated by BCL6B in GC cell lines
To gain insights into the molecular mechanisms of the tumour suppressive effect by BCL6B, BCL6B-modulated downstream target genes were characterised by cDNA microarray in BCL6B stably transfected AGS. When compared with empty vector-transfected cells, the antitumorigenesis effect by BCL6B was mediated by regulating important genes involved in apoptosis and proliferation pathways (see table 2 in online supplement). BCL6B increased the expression of pro-apoptotic genes including tumour necrosis factor receptor superfamily member 1A (TNFRSF1A; 2.5-fold), ataxia telangiectasia mutated homologue (ATM; 4.4-fold) and cyclin-dependent kinase inhibitor 1A (CDKN1A, p21; 1.6-fold) and decreased the expression of anti-apoptotic genes including vascular endothelial growth factor A (VEGFA; −1.8-fold) and S100 calcium binding protein A4 (S100A4; −2.0-fold). BCL6B also exerted an anti-proliferative effect by decreasing proliferation genes cyclin-dependent kinase 4 (CDK4; −13.8-fold) and plasminogen activator urokinase receptor (PLAUR; −2.3-fold). In addition, enhanced protein expression of ATM and p53 was demonstrated in AGS cells following BCL6B transfection by western blot analysis (figure 4C).
Frequent BCL6B methylation in primary GCs
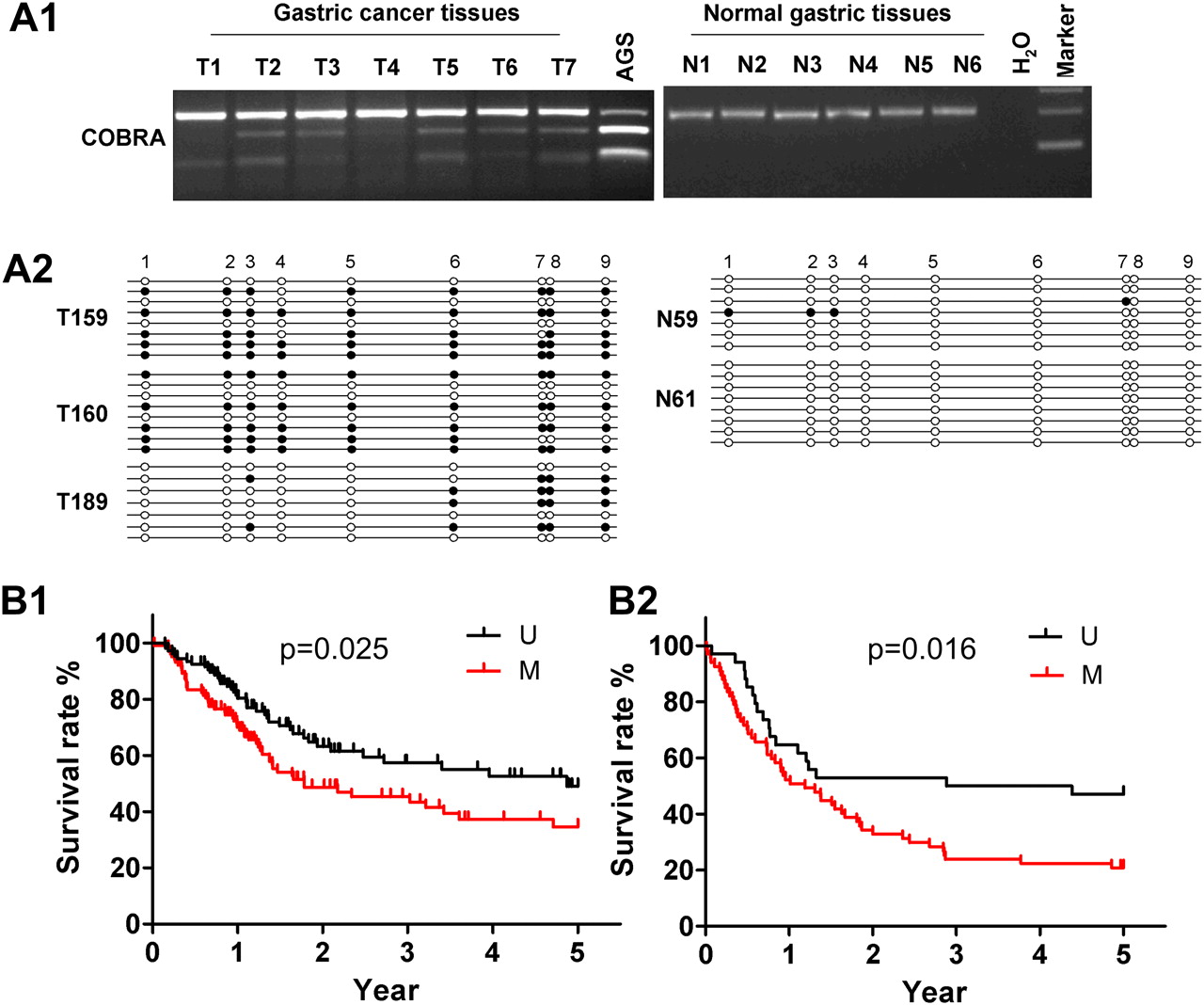
We further investigated BCL6B methylation status in 309 primary GCs from two independent cohorts and 20 normal gastric tissue specimens by COBRA. Frequent methylation was detected in GCs (102/208 (49%) for cohort I, and 67/101 (66%) for cohort II, table 1), but no methylation was detected in 20 normal gastric tissue specimens (figure 5A1), indicating that BCL6B methylation is tumour-specific. Detailed BGS analyses confirmed the findings by COBRA that the BCL6B promoter was frequently methylated in primary GC but not in normal gastric tissue (figure 5A2).

(A1) Promoter methylation of BCL6B in primary gastric cancer (GC) and normal gastric tissues determined by combined bisulfite restriction analysis (COBRA). (A2) Bisulfite genomic sequencing (BGS) analysis confirmed the methylation status of BCL6B in primary GC but not in normal gastric tissues. (B) Kaplan–Meier curves of two cohorts of patients with GC. Patients with BCL6B methylation (M) had significantly poorer survival than those without methylation (U) based on the log-rank test in (B1) cohort I (p=0.025) and (B2) cohort II (p=0.016).
BCL6B is an independent predictor of poor outcome in patients with GC
The association of BCL6B methylation status and clinicopathological features including clinical outcome was analysed in 309 patients with GC with known survival data from cohort I (median survival time 25.8 months, range 0.23–60) and cohort II (median survival time 33.7 months, range 0–60). There was no correlation between BCL6B methylation and clinicopathological features such as age, gender, Helicobacter pylori infection, histological type or pathological stage in cohort I, while methylation of BCL6B was found to be associated with Lauren type of GC in cohort II (table 1).
The characteristics of patients with GC related to the survival status are shown in table 3 in the online supplement. In univariate Cox regression analysis of cohort I, BCL6B methylation was associated with an increased risk of cancer-related death (RR 1.60, 95% CI 1.07 to 2.39; p<0.05). As expected, tumour stage was also a significant prognostic factor (p<0.0001). After adjustment for potential confounding factors, multivariate Cox regression analysis showed that BCL6B methylation was an independent predictor of poorer survival of patients with GC (RR 2.14, 95% CI 1.36 to 3.36; p=0.001, table 2). As shown in the Kaplan–Meier survival curves, patients with GC with BCL6B methylation had significantly shorter survival than others (p=0.025, log-rank test; figure 5B1).
Multivariate Cox regression analysis of potential poor prognostic factors for patients with gastric cancer in cohorts I and II
Cohort II was used to further validate the findings from cohort I. Univariate Cox regression analysis of cohort II similarly indicated that BCL6B methylation was associated with a significantly increased risk of cancer-related death (RR 1.92, 95% CI 1.13 to 3.25; p=0.01; see table 3 in online supplement). Tumour stage was also a significant predictor of outcome (p=0.006). In multivariate Cox regression analysis (table 2), BCL6B methylation was confirmed to be associated with poor survival of patients with GC (p=0.02). As shown in the Kaplan–Meier survival curves, patients with GC with BCL6B methylation had significantly shorter survival than others (p=0.016, log-rank test; figure 5B2).
Discussion
In this study we have shown for the first time that BCL6B is widely expressed in normal human tissues but was absent or downregulated in all nine GC cell lines investigated. The reduced expression is associated with promoter methylation as examined by promoter methylation analyses. Bisulfite sequencing of the promoter region of the BCL6B gene showed dense methylation in GC cell lines. The silencing of BCL6B can be reversed by pharmacological demethylation, inferring that methylation is the predominant mechanism for the inactivation of BCL6B in GC. As 5-Aza only leads to partial reactivation of BCL6B in SNU719 and NCI87 cells, we tested whether histone modification mediates BCL6B silencing in these cells by treatment with the histone deacetylase inhibitor TSA. Further restored expression of BCL6B was observed in SNU719 and NCI87 treated with 5-Aza together with TSA compared with 5-Aza only (figure 1D2), suggesting that histone modifications also play a role in the transcriptional silence of BCL6B.16 We also demonstrated that the protein expression of BCL6B was significantly decreased in primary GC tissues compared with their adjacent non-tumour tissues. COBRA and BGS results showed the promoter of BCL6B was densely methylated in primary GC tissues, whereas there was no methylation in normal gastric tissue. These results suggest that BCL6B could be a potential tumour suppressor and its downregulation could have some role in the development of GC.
The putative tumour suppressor function of BCL6B in human GC was further investigated by both in vitro and in vivo assays. Ectopic expression of BCL6B in the silenced AGS and BGC823 cells showed significant growth-suppressing effect by inhibition of cell viability and colony formation. Moreover, BCL6B enhanced both early and late apoptosis in AGS and BGC823 cells as determined by flow cytometry. The diminution of tumour growth in BCL6B re-expressed cells was also confirmed in nude mice. Collectively, these results indicate for the first time that BCL6B functions as a tumour suppressor in GC. It has been reported that BCL6B regulates differentiation into stages or lineages in a human erythroleukemia cell line11 and it is an important molecule in spermatogonial stem cell self-renewal.17 Overexpression of BCL6B induces apoptosis in the mouse embryonic fibroblast cell line NIH 3T3.13 Our findings have highlighted the importance of BCL6B as a potential tumour suppressor in GC.
We further revealed the molecular basis by which BCL6B exerts its tumour suppressor activity in GC by induction of apoptosis and control of cell proliferation using a cDNA microarray and western blot analysis. The enhanced apoptosis activity of BCL6B was found to be mediated through both intrinsic and extrinsic caspase-dependent pathways as well as the downregulation of anti-apoptosis regulators (S100A4 and VEGFA). We observed that caspase-9, an important intrinsic apoptosis pathway element, was significantly enhanced by BCL6B. Once activated, caspase-9 processes other effector caspases (caspase 3, caspase 7 and PARP) to initiate the intrinsic caspase cascade (figure 6).18 BCL6B also increased the expression of key genes mediating the extrinsic apoptosis pathway, including extracellular death receptors TNFRSF1A and downstream apoptosis executor caspase-8. The extrinsic apoptosis pathway begins from the interaction of tumour necrosis factor receptors (TNFRs) with their specific ligands and this pathway subsequently initiates apoptosis via activation of caspase-8 and downstream protein cascade leading to cell death (figure 6).19 20 Among the TNFRs, TNFRSF1A is one of the most popular receptors since its ligand, tumour necrosis factor α (TNFα), has a role in a wide range of biological activities associated with apoptosis in many types of cells.21 Both the intrinsic and the extrinsic pathways converge on downstream executioners, which are responsible for cell collapse and death (figure 6).22 Moreover, ectopic expression of BCL6B leads to the downregulation of S100A4 and VEGFA. S100A4 was reported to suppress apoptosis in pancreatic cancer23 and VEGFA has an important anti-apoptosis effect.24 25 Collectively, these results suggest that the anti-tumour growth effect of BCL6B may be partly related to the induction of apoptosis via various apoptosis-regulating pathways mediated by BCL6B (figure 6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proposed scheme for induction of cell apoptosis and inhibition of cell growth by BCL6B. BCL6B induces tumour cell apoptosis through cross-talk of both intrinsic and extrinsic apoptosis pathways. BCL6B also suppresses tumour cell proliferation through upregulation of p21 and downregulation of cyclin-dependent kinase 4 (CDK4) and plasminogen activator (PLAUR). The anti-tumorigenic function of BCL6B was also modulated by upregulating tumour suppressors ataxia telangiectasia mutated homologue (ATM) and p53, which are involved in the above anti-tumorigenic pathways.
The anti-proliferative effect caused by BCL6B in GC is mostly attributable to the upregulation of p21 and the downregulation of CDK4 and PLAUR. It has been indicated that p21 is a critical cyclinE/CDK2 and cyclin D/CDK4 inhibitor, mediating p53-dependent cell cycle G1 phase arrest.26 27 CDK4 binds to cyclin D, then activates retinoblastoma (Rb) family members and results in cell proliferation.28 PLAUR has been reported to increase cancer proliferation, invasion and metastasis.29 An increased level of PLAUR is strongly correlated with a poor prognosis and unfavourable clinical outcome.30 Thus, the suppression of cell proliferation by BCL6B could be a consequence of upregulation of p21 and downregulation of CDK4 and PLAUR (figure 6).
The anti-tumorigenic function of BCL6B was also contributed to the upregulation of ATM and p53, two well-known tumour suppressor genes. ATM plays a critical role in activating p53, inducing apoptosis and suppressing tumorigenesis.31 ATM kinase may directly, or indirectly through phosphorylation of CHK2, lead to phosphorylation and stabilisation of p53.32–35 Phosphorylation is a major post-translational modification of the DNA damage response pathway and has been shown to enhance protein stability and activity. Stabilisation and activation of p53 by ATM result in activation of a number of genes involved in the mediation of cell apoptosis and proliferation, such as p21 and caspase-9 (figure 6). Thus, upregulation of ATM and p53 by BCL6B also has a role in the anti-tumorigenic property of BCL6B.
To determine the clinical relevance of BCL6B in GC in vivo, we examined the promoter methylation of BCL6B by COBRA in 309 primary GCs from two cohorts of patients with GC and 20 normal controls. BCL6B gene promoter was found to be commonly methylated in GC tissues (49% in cohort I, 66% in cohort II) but not methylated in normal controls. It is thus worth further exploring the possible application of BCL6B tumour-specific promoter methylation as an epigenetic biomarker for patients with GC. The clinical outcome of GC varies greatly depending on the growth status and aggressiveness of individual tumours. TNM staging is still the most important clinical predictor of patient outcome. However, in many patients recurrence of GC occurs even at early stages. Additional prognostic biomarkers are therefore required to provide better risk assessment. Promoter methylation has been reported as a promising predictive biomarker in many human cancers.36 However, the contribution of epigenetic changes to the prognosis of GC is still largely unknown. Recognising the biological functions of BCL6B in GC, the inactivation of this gene by promoter methylation would favour tumour progression and a worse outcome. In this connection, the clinical significance of BCL6B promoter methylation and its associations with patient outcome was evaluated in two independent cohorts of 208 patients with GC in cohort I from Guangzhou, China and 101 patients in cohort II from Hong Kong. Our results indicated that promoter methylation of BCL6B was significantly associated with poorer survival in the patients with GC in cohort I independent of patient characteristics (RR 2.14, p=0.001). This finding was supported by the results of cohort II (RR 1.85, p=0.02). Our data support an adverse effect of loss of BCL6B expression regulated by promoter methylation on survival of patients with GC, providing additional evidence for the role of BCL6B as a novel candidate tumour suppressor gene in the development of GC.
In conclusion, we have identified a novel functional tumour suppressor gene, BCL6B, inactivated by promoter methylation in GC. BCL6B induced tumour cell apoptosis through cross-talk of both intrinsic and extrinsic apoptosis pathways. BCL6B suppressed tumour cell proliferation through upregulation of p21 and downregulation of CDK4 and PLAUR. The anti-tumorigenic function of BCL6B was also modulated by upregulating tumour suppressors ATM and p53, which are involved in the above anti-tumorigenic pathways. BCL6B methylation may serve as a potential epigenetic biomarker to predict the outcome of patients with GC.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
LX and XL contributed equally to the paper.
Funding This project was supported by National Natural Science Foundation of China (81172346), Research Grants Council RGC CERG CUHK (473008 and 465808), Group Research Scheme CUHK (3110043), CUHK Focused Investment Gant (2041423, 1903026), National Natural Science Foundation of China (81172346) and RFCID (10090942).
Competing interests None.
Patient consent Obtained.
Ethics approval The study protocol was approved by the Clinical Research Ethics Committee of the Sun Yat-sen University of Medical Sciences and the Ethics Committee of the Chinese University of Hong Kong.
Provenance and peer review Not commissioned; externally peer reviewed.