Article Text

Abstract
Objective Vascular remodelling during liver damage involves loss of healthy liver sinusoidal endothelial cell (LSEC) phenotype via capillarisation. Hedgehog (Hh) signalling regulates vascular development and increases during liver injury. This study therefore examined its role in capillarisation.
Design Primary LSEC were cultured for 5 days to induce capillarisation. Pharmacological, antibody-mediated and genetic approaches were used to manipulate Hh signalling. Effects on mRNA and protein expression of Hh-regulated genes and capillarisation markers were evaluated by quantitative reverse transcription PCR and immunoblot. Changes in LSEC function were assessed by migration and tube forming assay, and gain/loss of fenestrae was examined by electron microscopy. Mice with acute or chronic liver injury were treated with Hh inhibitors; effects on capillarisation were assessed by immunohistochemistry.
Results Freshly isolated LSEC expressed Hh ligands, Hh receptors and Hh ligand antagonist Hhip. Capillarisation was accompanied by repression of Hhip and increased expression of Hh-regulated genes. Treatment with Hh agonist further induced expression of Hh ligands and Hh-regulated genes, and upregulated capillarisation-associated genes; whereas Hh signalling antagonist or Hh ligand neutralising antibody each repressed expression of Hh target genes and capillarisation markers. LSEC isolated from SmoloxP/loxP transgenic mice that had been infected with adenovirus expressing Cre-recombinase to delete Smoothened showed over 75% knockdown of Smoothened. During culture, Smoothened-deficient LSEC had inhibited Hh signalling, less induction of capillarisation-associated genes and retention of fenestrae. In mice with injured livers, inhibiting Hh signalling prevented capillarisation.
Conclusions LSEC produce and respond to Hh ligands, and use Hh signalling to regulate complex phenotypic changes that occur during capillarisation.
- Alcoholic liver disease
- basic sciences
- cancer
- carcinogen metabolism
- cell biology
- cell signalling
- cirrhosis
- fatty liver
- fibrogenesis
- fibrogenesis
- fibrosis
- hepatic stellate cell
- hepatocellular carcinoma
- liver
- liver immunology
- liver regeneration
- molecular mechanisms
- non-alcoholic steatohepatitis
Statistics from Altmetric.com
- Alcoholic liver disease
- basic sciences
- cancer
- carcinogen metabolism
- cell biology
- cell signalling
- cirrhosis
- fatty liver
- fibrogenesis
- fibrogenesis
- fibrosis
- hepatic stellate cell
- hepatocellular carcinoma
- liver
- liver immunology
- liver regeneration
- molecular mechanisms
- non-alcoholic steatohepatitis
Significance of this study
What is already known about this subject?
-
Liver injury induces capillarisation of LSEC and results in remodelling of the hepatic vasculature.
-
The Hh pathway regulates vascular development during embryogenesis.
-
Liver injury triggers activation of the Hh pathway.
What are the new findings?
-
Freshly isolated LSEC express both Hh ligands and receptors, and activation of Hh signalling is accompanied by LSEC capillarisation in vitro.
-
Cultured LSEC respond to Hh agonist, antagonist and Hh ligand neutralising antibody, thus are Hh-responsive cells.
-
LSEC capillarisation can be prevented by the inhibition of the Hh pathway both in vitro and in vivo.
How might it impact on clinical practice in the foreseeable future?
-
Our data suggest a novel way to prevent and/or reverse LSEC capillarisation, a process involved in liver fibrosis progression.
The liver sinusoidal endothelial cell (LSEC) has a unique phenotype among all mammalian endothelial cells, ie, LSEC have open fenestrae grouped into sieve plates and lack an organised basement membrane. LSEC lose this highly specialised morphology during a process called capillarisation. Capillarisation occurs in vivo during many different kinds of liver injury (eg, fibrosis,1 ,2 hepatitis,3 ,4 alcoholic liver injury,5 arsenic poisoning)6 and increases naturally with age (called pseudo-capillarisation).7 The process can be modelled in vitro by culturing LSEC on plastic dishes in serum-containing medium.8 ,9 However, the molecular mechanisms driving capillarisation have not been fully elucidated. Improved understanding of the latter will clarify how LSEC maintain their unique phenotype and identify therapeutic targets to retain and/or restore their healthy phenotype during liver injury or ageing.
During fetal development, angiogenesis and vasculogenesis are regulated partly by the Hedgehog (Hh) pathway.10 ,11 Therapeutic activation of this pathway has also been reported to improve vascularisation of injured tissues in adults.12 ,13 These findings demonstrate that certain types of endothelial cells are Hh responsive, and prompted us to investigate the hypothesis that Hh is one of the factors that controls capillarisation. Hh is a conserved morphogenic signalling pathway that modulates the fates of various types of cells, including endothelial progenitors.14 ,15 In Hh-responsive cells, the canonical Hh signalling pathway is activated when any of the three Hh ligands (Sonic hedgehog (Shh), Indian hedgehog (Ihh) or Desert hedgehog) engage the plasma membrane spanning receptor, Patched (Ptc). This interaction prevents Ptc from repressing Smoothened (Smo), a co-receptor-like molecule that transduces Hh-initiated signalling intracellularly. The latter eventually result in the stabilisation and nuclear localisation of Gli-family transcription factors (Gli1, Gli2 and Gli3). Gli-binding, in turn, regulates the transcriptional activity of Hh target genes that influence cell viability, proliferation and differentiation.16 ,17
Canonical Hh pathway activity is very low in healthy adult livers because Hh ligands are scarce. However, liver injury increases local production of Hh ligands. This promotes Hh pathway activation, and thus Hh signalling becomes dramatically activated in damaged livers.18–20 Several cell types that accumulate in injured livers, including myofibroblastic hepatic stellate cells (HSC), various immune cells and different types of liver progenitor cells, produce and respond to Hh ligands. Genetic and pharmacological approaches that modulate Hh pathway activity have been shown to influence liver fibrosis, regeneration and cancer. There is thus growing evidence that Hh signalling plays a significant role in the adult liver repair.21 ,22
A few reports suggest that Hh signalling may regulate vascular remodelling responses to liver injury.18 ,23 For example, similar to cultured human vascular endothelial cells,24 cultured LSEC respond to exogenous Hh ligands by increasing the expression of Hh target genes. The Hh-regulated transcription factor, Gli2, has also been demonstrated in nuclei of LSEC within injured liver tissue using immunohistochemistry.18 ,23 However, knowledge about how Hh pathway activation influences the phenotype of LSEC is quite limited. Improved understanding of this issue will clarify whether or not Hh pathway activation promotes capillarisation of hepatic sinusoids during liver injury and ageing. Therefore, we examined LSEC at different time points during culture-provoked capillarisation to profile changes that might have resulted from Hh pathway activation, and then used genetic, antibody-mediated and pharmacological approaches to manipulate Hh signalling to determine if, and how, this modified the capillarisation process.
Materials and methods
Reagents
Chemicals were obtained from Sigma-Aldrich Corporation (St Louis, Missouri, USA) unless stated otherwise. Reagents used include vascular endothelial growth factor (VEGF; Peprotech, Rocky Hill, New Jersey, USA), osmium tetroxide, glutaraldehyde, cacodylate buffer, tannic acid (Electron Microscopy Sciences, Hatfield, Pennsylvania, USA), hexamethyldisilazane (Ted Pella, Redding, California, USA), cyclopamine and tomatidine (Toronto Research Chemicals, Toronto, Canada), SAG (Enzo Life Sciences, Plymouth Meeting, Pennsylvania, USA), 5E1-neutralising antibody (Iowa Hybridoma Bank, University of Iowa, Iowa, USA), GDC-0449 (Selleck Chemicals, Houston, Texas, USA), mouse IgG1 isotype control antibody (R&D Systems, Minneapolis, Minnesota, USA), Dil-Ac-LDL (Biomedical Technologies, Stoughton, Massachusetts, USA), iodixanol (Accurate Chemical and Scientific, Westbury, New York, USA), phycoerythrin-conjugated mouse anti-rat CD31 (BD Pharmingen, San Diego, California, USA), rabbit polyclonal antibody to desmin (Abcam, Cambridge, Massachusetts, USA), CD105 and F4/80 antibody (BioLegend, California, USA), endothelin-1 antibody (Thermo Fisher Scientific, Rockford, Illinois, USA), inducible nitric oxide synthase (iNOS) antibody (Santa Cruz Biotechnology, Santa Cruz, California, USA). FITC-labelled formaldehyde-treated serum albumin (FITC–FSA) was prepared as previously described.25
Animals
Male Sprague–Dawley rats weighing 200 g were obtained from Charles River Laboratories (Wilmington, Massachusetts, USA). SmoloxP/loxP transgenic mice26 and C57BL/6 mice were obtained from the Jackson Laboratory (Bar Harbor, Maine, USA). Mdr2−/− mice were a gift from Dr Detlef Schuppan (Beth Israel Deaconess Medical Center, Boston, Massachusetts, USA). GDC-0449 treatment,27 70% partial hepatectomy and cyclopamine treatment21 were performed as previously described. Animal experiments fulfilled National Institutes of Health and Duke University Institutional Animal Care and Use Committee requirements for humane animal care.
LSEC isolation
LSEC were isolated from normal Sprague–Dawley rats or SmoloxP/loxP transgenic mice by collagenase perfusion, iodixanol density gradient centrifugation and centrifugal elutriation as previously described28 with modification. Yields of LSEC are on average 80 million sinusoidal endothelial cells per 10 g liver with viability of more than 95% and purity greater than 96%, as determined by scanning electron microscopy (SEM), fluorescence-activated cell sorter (FACS) for CD31 and CD105, and positive staining for fluorescent acetylated low-density lipoprotein (Dil-Ac-LDL; Biomedical Technologies) and FITC–FSA. FACS on F4/80 and desmin were used to exclude contamination by macrophage/Kupffer cells and HSC. Cells were cultured on rat-tail collagen-coated plates (400 000 cells/cm2) in Dulbecco's minimal essential medium low glucose with 10% fetal bovine serum (FBS). Cells usually attached to the culture dish within 3 h of plating.
Pharmacological manipulation of Hh signalling
LSEC were treated with Smo antagonist, cyclopamine (3 μM), or tomatidine, a catalytically inactive analogue (3 μM), for 48 h.19 In separate experiments, Hh-neutralising antibody (5E1, 10 μg/ml) or control IgG were added to the LSEC culture.29 SAG, an Hh agonist (0.3 μM), was used to activate the Hh pathway.30
Adenoviral transduction of LSEC
Primary LSEC isolated from SmoloxP/loxP transgenic mice (n=10) were cultured in the presence of adenoviral vectors carrying green fluorescent protein (Ad-GFP) or Cre recombinanse (Ad-Cre) (multiplicity of infection 25).31 Cultures were analysed at 48 h. In separate experiments, SmoloxP/loxP mice were infected with Ad-GFP (50 μl) or Ad-Cre by tail vein injection. LSEC were isolated 2 days later and cultured on collagen-coated plates for an additional 2 days before further analysis.
SEM and quantitative imaging
Sample preparation was performed as previously described.32 Briefly, LSEC and livers were fixed with glutaraldehyde, postfixed with osmium tetroxide, dehydrated with graded alcohols, dried with hexamethyldisilazane, sputter-coated with gold and examined using a FEI XL30 ESEM scanning electron microscope (FEI, Hillsboro, Oregon, USA). Porosity (the percentage of LSEC surface occupied by fenestrae) was measured in SEM micrographs of cells;32 in brief, the total LSEC surface area and the open area of individual fenestrae were quantified. Open areas were summed, divided by the total surface area, expressed as a percentage of the open area and averaged. Each average was taken from 15 images and analysis was done using MetaMorph software (Molecular Devices, Downingtown, PA, USA).
mRNA quantification by real-time reverse transcription PCR
Total RNA was prepared using RNeasy Mini Kit (Qiagen, Valencia, California, USA) according to the manufacturer's instruction; 1.5 μg of RNA was reverse-transcribed using random primers and Superscript RNase H-reverse transcriptase (Invitrogen, Carlsbad, California, USA). Samples were incubated at 25°C for 15 min, 42°C for 55 min; reverse transcriptase was inactivated by heating at 70°C for 15 min followed by cooling at 4°C for 10 min. Complementary DNA samples were used for quantitative reverse transcription PCR (qRT–PCR) using iQ-SYBR Green Supermix (Bio-Rad Laboratories, Hercules, California, USA).19 β-Actin was used as internal control. For qRT–PCR parameters were as follows: denaturating at 95°C for 3 min, followed by 40 cycles of denaturing at 95°C for 10 s and annealing extension at the optimal primers temperatures for 60 s. Threshold cycles were automatically calculated by the iCycler iQ real-time detection system. Target gene levels in the cells are presented as a ratio to levels detected in the corresponding control cells according to the ΔΔCt method. Primer sequences are listed in supplementary tables 1 and 2 (available online only).
Western blotting
Western blotting was performed as previously described;19 results were normalised to β-actin expression.
Migration assay
Migration was measured by using a modified Boyden chamber assay. LSEC, 400 000 cells/cm2, were plated on collagen-coated Transwell inserts with 8-μm pore size (Costar, Corning, New York, USA) in Dulbecco's minimal essential medium low glucose supplemented with 2% FBS. Inserts were placed on top of 2 cm2 wells containing medium plus 2% FBS with different treatment. Cells migrated were counted after 20 h in 10 random 20× fields for each insert.
Tube formation assay
LSEC were seeded on growth factor-reduced Matrigel (BD Bioscience, Bedford, MA, USA) with/without treatment as described.18 The length of capillary-like tube formation was quantified in 10 randomly chosen optical fields using light microscopy after 6 h.
Immunohistochemistry
Formalin-fixed, paraffin-embedded samples were deparaffinised and rehydrated. Antigen retrieval was performed by heating in 10 mM sodium citrate buffer. Sections were blocked (Dako Envision, Carpinteria, California, USA) and incubated with Gli2 antibody (Genway, San Diego, California, USA) overnight at 4°C and developed with diaminobenzidine, followed by incubation with CD31 antibody (Santa Cruz Biotechnology) overnight at 4°C and developed with Ferangi blue (Biocare Medical, Concord, California, USA). The number of Gli2/CD31 double-positive cells were counted in five representative 63× fields/section from each sample (n=3).
Statistics
All data, expressed as mean±SEM, were from at least three separate experiments. Groups were compared by analysis of variance with à posteriori contrast by least significant difference; or by Student's t test using the Microsoft Excel Analysis ToolPak (Microsoft, Redmond, Washington, USA). Results with p<0.05 were considered significant.
Results
Characterisation of elutriated primary LSEC
LSEC isolated using cell elutriation methodology demonstrate typical morphology under light microscopy (figure 1A). SEM confirms that they possess fenestrae grouped into sieve plates (figure 1B), the defining feature of the liver sinusoidal endothelium.28 By flow cytometry, approximately 96% of these cells are double-positive for two endothelial markers, CD31 and CD105 (endoglin),33 ,34 whereas less than 2% express the macrophage marker, F4/80, and none are positive for desmin, a marker of quiescent and myofibroblastic HSC (figure 1C). The aggregate findings, therefore, indicate that our primary LSEC preparations are a highly pure population. The vast majority of these cells were able to take up Dil-Ac-LDL (figure 1D) and FITC–FSA (figure 1E), demonstrating that they remain endocytically active in vitro. Furthermore, on day 5, most of the cells were still able to take up Dil-Ac-LDL (see supplementary figure 1A, available online only), but not FITC–FSA (see supplementary figure 1B, available online only), consistent with previous reports,9 ,35 suggesting that LSEC lose their endocytic activity during culture.

Characterisation of liver sinusoidal endothelial cells isolated by the elutriation method. (A) Liver sinusoidal endothelial cell (LSEC) cultured for 1 day on collagen-coated plate demonstrate typical ‘cobblestone’ morphology under phase contrast microscopy. Scale bar: 100 μm. (B) Scanning electron microscopy (SEM) shows LSEC have fenestrae grouped into sieve plates (arrow), the defining morphology of LSEC. Scale bar: 5 μm. (C) Fluorescence-activated cell sorter analysis of freshly isolated LSEC. Approximately 96% of LSEC were double-positive for CD31 and CD105 (endoglin). Only 2% of LSEC were F4/80 positive (Kupffer cell marker) and none expressed desmin (hepatic stellate cell marker). Uptake of fluorescent acetylated low-density lipoprotein (Ac-Di-LDL) (D) and FITC-labelled formaldehyde-treated serum albumin (FSA) (E) by day 1 LSEC demonstrate the typical endocytic activity of the cells. Scale bar: 50 μm for left images, 20 μm for right images. All experiments were performed at least three times.
Primary LSEC undergo capillarisation in vitro
It is well established that LSEC lose their healthy, quiescent phenotype (fenestrae grouped into sieve plates) rapidly during culture.8 ,36 During the first day of culture, our LSEC maintained normal fenestrae and sieve plates, but both of these features were dramatically reduced after 2 or 3 days of culture. No sieve plates were observed, and only occasional fenestrae remained, by culture days 4 or 5 (figure 2A). The loss of macroscopically evident fenestrae was further confirmed by porosity measurement (figure 2A). As others have reported, the morphological changes in LSEC that occurred during culture-induced capillarisation were accompanied by the downregulation of messenger RNA for endothelial nitric oxide synthase,37 VEGF receptor 1/2,38 and upregulation of mRNA and protein for iNOS23 and endothelin 139 (figure 2B,C). Interestingly, mRNA and protein expression of CD31 (figure 2B,C), a commonly- used marker for LSEC capillarisation,6 ,8 was easily demonstrated in our freshly isolated LSEC. CD31 mRNA levels fell by approximately 80% during the initial day of culture, but eventually recovered to approximately 50% of basal levels by day 3. Earlier studies typically compared CD31 expression between day 1 and day 3 LSEC, and noted an increase in CD31 protein/mRNA during this time interval.8 ,40 Therefore, our findings are consistent with those reports. Our western blot analysis demonstrated that whole cell levels of CD31 protein remained relatively constant during LSEC culture, mirroring similar data from Géraud et al.38 Because LSEC expression of CD31 remains relatively constant in this model of capillarisation, we used changes in mRNA and protein levels of iNOS, another capillarisation marker (figure 2B,C), to track capillarisation in subsequent experiments.

Liver sinusoidal endothelial cells (LSEC) undergo capillarisation spontaneously in vitro. (A) Rat LSEC were cultured on collagen-coated plates, processed and examined by scanning electron microscopy at ×10 000 magnification. Day 1 LSEC show numerous fenestrae grouped into sieve plates (arrow). Days 2 and 3 LSEC have fewer fenestrae and sieve plates (arrow). Days 4 and 5 LSEC have only occasional fenestrae (arrowhead). Porosity measurement was used to quantify the percentage of fenestrae per surface area. Scale bar: 5 μm. **p<0.01 day 1 cells, n=3. (B) Quantitative reverse transcription PCR analysis of endothelial cell-associated gene expression changes during culture-induced capillarisation. *p<0.05, **p<0.01, ***p<0.001 versus day 0 cells, n=3. (C) Western blot analysis of protein harvested from rat LSEC with densitometry to confirm the gene expression change described in (B). Results are representative of triplicate experiments. *p<0.05, **p<0.01 versus day 0 cells, #non-detectable. ET-1, endothelin 1; eNOS, endothelial nitric oxide synthase; iNOS, inducible nitric oxide synthase; VEGFR, vascular endothelial growth factor receptor.
Primary LSEC produce and express Hh target genes
mRNA and protein expression of Shh ligand, Hh signalling components and various Hh target genes was assessed in freshly isolated LSEC and at different time points during culture-induced capillarisation. Freshly isolated LSEC express Shh ligand, components of the canonical Hh signalling pathway (eg, the receptor Ptc, co-receptor Smo and Hh-regulated transcription factors Gli2 and Gli3), and several Hh target genes (eg, Hhip, Ptc and sFRP1) (figure 3A and B). During the first day of culture, Hh interacting protein (Hhip, a Hh ligand antagonist) expression virtually disappears at both the mRNA and protein level and there is increased accumulation of Shh protein (figure 3A,B). This is accompanied by increased mRNA expression of the Hh-regulated transcription factors, Gli2 and Gli3 (figure 3A). Day 1 LSEC maintain normal fenestrae (figures 1B and 2A), thus capillarisation is not evident in day 1 LSEC. Therefore, activation of Hh signalling occurs before the first overt evidence of capillarisation (loss of fenestrae), which emerges on day 2 (figure 2A). As capillarisation progresses, there is further upregulation of both Gli2 and Gli3 during culture days 3–5 (figure 3A and B), indicating increased Hh pathway activity in capillarised LSEC compared with freshly isolated LSEC. Increased mRNA expression of several genes that are known to be transcriptionally activated by Gli2 (eg, Ptc, soluble frizzled-related peptide 1 (sFRP1),41 osteopontin30 and the mesenchymal marker, Twist, figure 3A,B) occurred when LSEC exhibited increased accumulation of Gli2 and Gli3 proteins.

Hedgehog (Hh) pathway is activated during liver sinusoidal endothelial cell (LSEC) capillarisation in vitro. (A) Rat LSEC were cultured, mRNA were harvested and quantitative reverse transcription PCR were used to analyse the expression change of Hh target genes (Gli2, Gli3, Sonic Hh (Shh), Hedgehog-interacting protein (Hhip), Patched (Ptc), Smoothened, Twist2, secreted frizzled-related protein 1 (sFRP1), osteopontin (OPN)). *p<0.05, **p<0.01, ***p<0.001 versus day 0 cells, n=3. (B) Western blot analysis of protein harvested from rat LSEC with densitometry to confirm the gene expression change described in (B). β-Actin was used as loading control. Results are representative of triplicate experiments. *p<0.05, **p<0.01, ***p<0.001 versus day 0 cells, #non-detectable.
LSEC are Hh-responsive cells during culture
To examine the significance of Hh signalling during capillarisation, Hh pathway activity was manipulated in day 3 LSEC (which are capillarised, figure 2A). Forty-eight hours following further activation of Hh signalling by treatment with a Hh agonist (SAG, 0.3 μM), we noted a twofold or greater increase in mRNA encoding Hh ligands (Shh, Ihh), Hh-sensitive transcription factors (Gli2, Gli3) and Hh-target genes (sFRP1, osteopontin). Augmenting Hh pathway activity also caused LSEC to upregulate further their expression of activation markers (eg, iNOS) and mesenchymal genes (eg, snail, twist) (figure 4A). Conversely, inhibition of endogenous Hh signalling by treating LSEC with a Smoothened antagonist (cyclopamine, 3 μM) resulted in greater than 50% repression of Hh ligands, Hh signalling components, Hh target genes and LSEC activation markers (figure 4B). Treating LSEC with Hh ligand neutralising antibody (5E1, 10 μg/ml) resulted in similar changes in target gene expression (figure 4C), suggesting an autocrine effect of Hh ligands in activating Hh signalling in capillarised LSEC.

Liver sinusoidal endothelial cells (LSEC) are Hedgehog (Hh) responsive. (A) Rat LSEC were cultured for 3 days and treated with either dimethyl sulphoxide or SAG (0.3 μM, a Hh agonist) for two more days. mRNA were isolated and gene expression changes were examined by quantitative reverse transcription (qRT)–PCR. (B) Day 3 rat LSEC were treated with either cyclopamine (3 μM, a pharmacological inhibitor of Hh signalling) or tomatidine (an inert cyclopamine analogue) for 2 days. mRNA was harvested, and changes in gene expression were determined by qRT–PCR. (C) Rat LSEC were cultured for 3 days and treated with either 5E1 (10 μg/ml, Hh ligand neutralising antibody) or IgG for two more days. mRNA was isolated and gene expression changes were monitored by qRT–PCR. *p<0.05, **p<0.01, ***p<0.001, n=3. ET-1, endothelin 1; iNOS, inducible nitric oxide synthase; OPN, osteopontin; sFRP1, secreted frizzled-related protein 1.
To exclude the possibility that these gene expression changes were caused by potential off-target effects of cyclopamine,42 LSEC were isolated from SmoloxP/loxP transgenic mice, cultured for 3 days, infected with adenovirus-expressing Cre-recombinase (Ad-Cre, for conditional deletion of Smo), and harvested 48 h later (on day 5 of culture). This approach knocked down Smo gene expression by 76.59±3.60%, and caused 40–50% repression of Shh and various Hh target genes (eg, Gli1, Gli2), as assessed by qRT–PCR and/or western blot (figure 5A,B). Targeted disruption of Smo gene expression also downregulated the expression of iNOS, CD31 and endothelin 1, supporting a role for the Hh pathway in maintaining the phenotype of capillarised LSEC.

Effect of knocking down liver sinusoidal endothelial cell (LSEC) Smo gene expression in vitro on the Hedgehog (Hh) pathway. (A) Primary LSEC were isolated from SmoloxP/loxP transgenic mice, placed in monocultures for 3 days, and infected with either adenovirus expressing green fluorescent protein (Ad-GFP, control) or Cre-recombinase (Ad-Cre, for conditional deletion of the Hh signalling intermediate, Smo). After 48 h, cells were harvested to obtain mRNA for quantitative reverse transcription PCR analysis. *p<0.05, **p<0.01, ***p<0.001 versus control. n=3. (B) Western blot analysis of protein harvested from mouse LSEC described in (A) with densitometry to confirm the gene expression change. β-Actin was used as loading control. *p<0.05, **p<0.01 versus control. Results are representative of triplicate experiments. ET-1, endothelin 1; iNOS, inducible nitric oxide synthase.
Hh signalling promotes LSEC migration and vascular tube formation
It is known that Hh pathway activation can induce migration and capillary tube formation by different types of cultured endothelial cells,11 ,24 ,43–45 including LSEC.18 To explore further the role of Hh signalling on migration and vascular tube formation by LSEC, Hh pathway activity was manipulated in isolated LSEC cultured on collagen-coated inserts or matrigel. Inhibiting Hh signalling with cyclopamine impaired the ability of LSEC to migrate (figure 6C,F) and form tubes in vitro (figure 7C,F), whereas activation of the Hh pathway by SAG promoted both migration (figure 6E,F) and vascular tube formation (figure 7E,F). Cyclopamine also blocked the ability of VEGF to induce LSEC migration (figure 6D,F) and tube formation (figure 7D,F), possibly by inhibiting VEGF receptor 1 and VEGF receptor 2 expression by LSEC (see supplementary figure 2, available online only). Collectively, these data confirmed that Hh signalling promotes complex phenotypic changes in LSEC that are necessary for them to migrate and form vascular tubes in vitro.
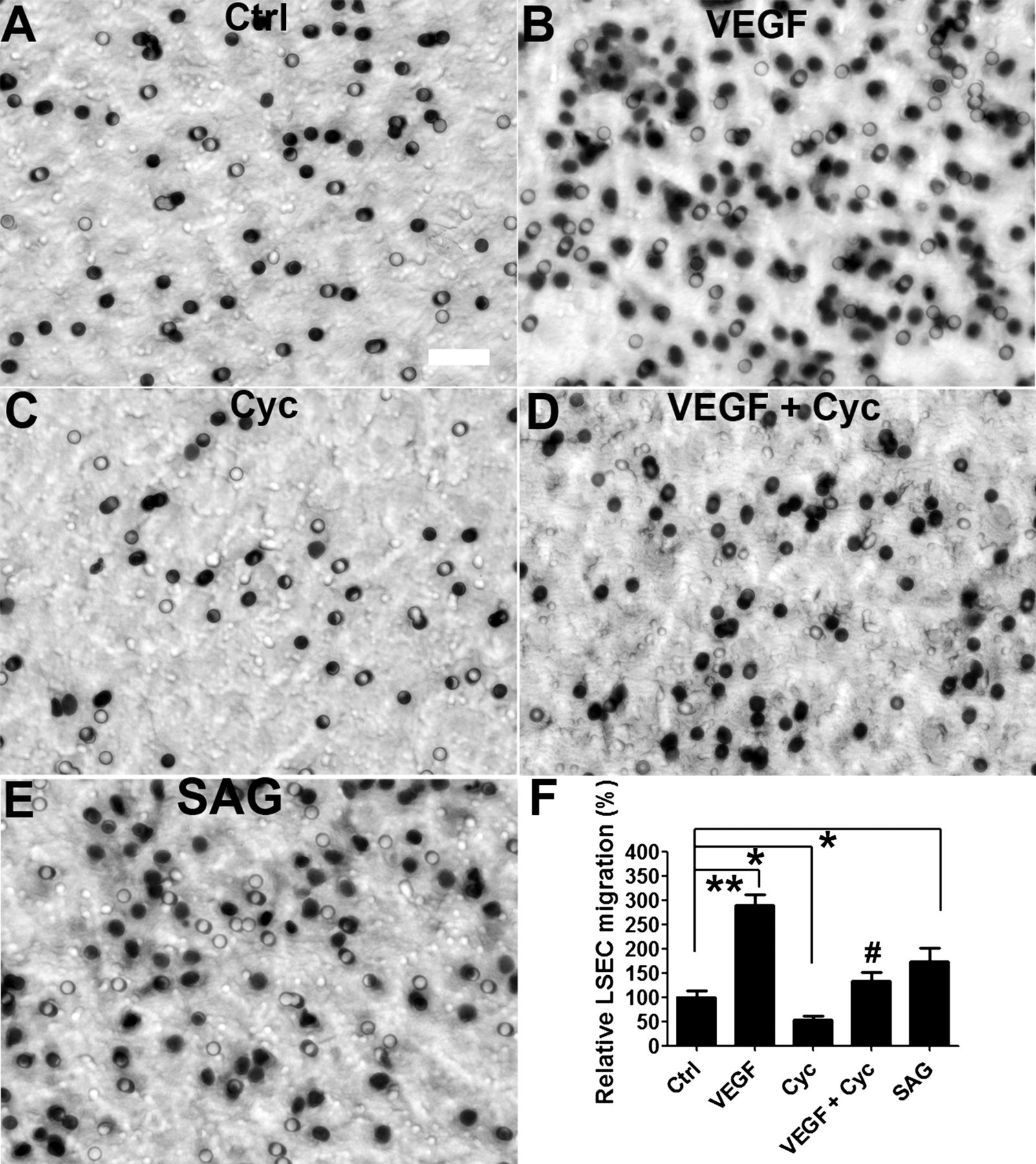
The Hedgehog (Hh) pathway promotes liver sinusoidal endothelial cell (LSEC) migration in vitro. Primary rat LSEC were seeded on collagen-coated insert and treated with (A) normal growth medium (ctrl), (B) 40 ng/ml vascular endothelial growth factor (VEGF), (C) 3 μM cyclopamine (Cyc), (D) VEGF with cyclopamine (VEGF + Cyc), or (E) 0.3 μM SAG for 20 h. (F) Effects on migration were assessed by counting the number of cells in the bottom of the inserts in triplicate experiments. Representative micrographs and statistical summary are shown. *p<0.05, **p<0.01 versus control. #p<0.05 versus VEGF. Scale bar: 50 μm.

The Hedgehog (Hh) pathway promotes liver sinusoidal endothelial cell (LSEC) tube formation in vitro. Primary rat LSEC were seeded on growth factor-reduced Matrigel and treated with (A) normal growth medium (ctrl), (B) 40 ng/ml vascular endothelial growth factor (VEGF), (C) 3 μM cyclopamine (Cyc), (D) VEGF with cyclopamine (VEGF + Cyc), or (E) 0.3 μM SAG for 6 h. (F) Effects on vascular tube formation were assessed by quantifying the length of capillary-like tube in triplicate experiments. Representative micrographs and statistical summary are shown. *p<0.05 versus control. #p<0.05 versus VEGF. Scale bar: 100 μm.
Hh signalling regulates LSEC capillarisation
LSEC become capillarised after 2 days culture (figure 2A), and this process is accompanied by activation of the Hh pathway (figure 3A,B). Inhibiting Hh signalling in capillarised LSEC by treating them with cyclopamine or anti-Hh neutralising antibody significantly repressed the expression of capillarisation-associated genes, such as iNOS, CD31 and endothelin 1 (figure 4B,C). Similar effects were achieved by knocking down Smo gene expression in culture-capillarised LSEC (figure 5A,B). However, SEM examination demonstrated that none of these approaches restored LSEC fenestrae (data not shown). These data suggest, therefore, that once LSEC become capillarised, inhibiting Hh signalling only partly reverses their capillarised phenotype.
To determine whether capillarisation might be prevented by blocking the induction of Hh signalling in LSEC before capillarisation begins, freshly isolated, primary LSEC were immediately incubated with cyclopamine or an inactive cyclopamine analogue (control) after attachment to culture plates. LSEC treated with the control agent underwent capillarisation, completely losing fenestrae within 2 days after plating. In contrast, LSEC that were treated with cyclopamine immediately after plating maintained porosity (figure 8A,B). LSEC cultures that retained the fenestrated phenotype exhibited less induction of mRNA that mark Hh signalling (Gli2) and capillarisation (iNOS) (figure 8C). Because the process of LSEC isolation per se might initiate the cascade of events that culminates in capillarisation, we performed further studies in which we abrogated Hh signalling in LSEC before the cells were isolated. This was accomplished by injecting adenoviral vectors for Cre recombinase (Ad-Cre) into SmoloxP/loxP mice 48 h before liver perfusion and LSEC harvest. Results were compared with SmoloxP/loxP mice that were similarly treated with an irrelevant adenovirus encoding green fluorescent protein (Ad-GFP). LSEC from the mice that were injected with Ad-Cre virus in vivo exhibited knockdown of smo, and smo-depleted LSEC failed to upregulate the expression of Shh ligand, Gli2, or iNOS after 2 days in culture (figure 8D). The inhibited induction of Hh signalling and capillarisation-associated genes in the smo-deficient LSEC was accompanied by the maintainenance of fenestrae (figure 8E,F), providing further evidence that inhibition of Hh signalling prevents LSEC from undergoing capillarisation.

Inhibition of Hedgehog (Hh) pathway prevents liver sinusoidal endothelial cell (LSEC) capillarisation in vitro. Primary rat LSEC were plated on collagen-coated coverslip. After 3 h, LSEC attached and were then incubated with either tomatidine (Tom) or cyclopamine (Cyc, 3 μM). Cells were fixed and processed for scanning electron microscopy (SEM) on day 2. Representative (A) SEM micrograph and (B) porosity measurement were shown. LSEC treated with cyclopamine maintained fenestrae grouped into sieve plates (circle), while LSEC treated with tomatidine lose fenestrae. (C) mRNA were also isolated from the treated cells described in (A) for quantitative reverse transcription (qRT)–PCR analysis on Gli2 and inducible nitric oxide (iNOS) gene expression. SmoloxP/loxP transgenic mice were pretreated with adenovirus expressing green fluorescent protein (Ad-GFP) or Cre (Ad-Cre) by tail vein injection. Primary LSEC were isolated 2 days after treatment and plated on a collagen-coated dish for another 2 days. (D) mRNA expression of Smo, Gli2, Shh and iNOS were examined by qRT–PCR. Cells were also fixed for (E) SEM micrograph and (F) porosity measurement was shown. Ad-Cre-infected LSEC maintain normal fenestrae grouped into sieve plates (circle), while Ad-GFP-infected LSEC lose fenestrae. Note that occasional fenestrae and sieve plates (arrow) can still be observed in Ad-GFP-infected LSEC, indicating that the loss of fenestrae was not caused by the contamination of other kinds of endothelial cells. *p<0.05, **p<0.01, ***p<0.001, n=3–6. Scale bar: 5 μm.
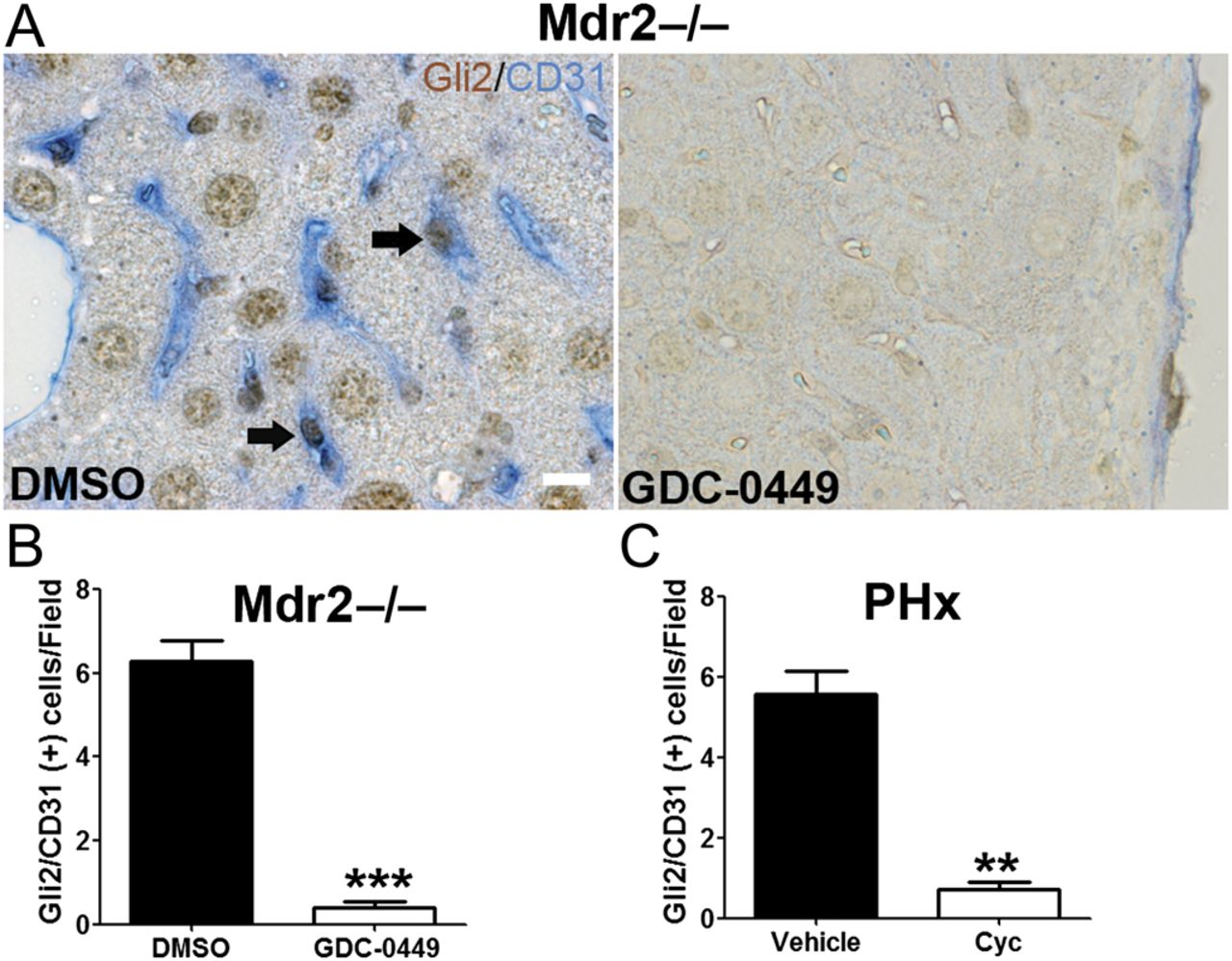
To validate that Hh signalling regulates LSEC capillarisation in vivo, Hh pathway activity was manipulated in different liver injury models: mdr2−/− mice27 (a model of chronic liver injury and repair that results in progressive biliary-type fibrosis) and wild-type mice following acute partial hepatectomy (a model of acute liver cell loss and repair). Mdr2−/− mice consistently expressed Hh ligands and progressively accumulated Hh-responsive liver myofibroblasts.27 Treatment of Mdr2−/− mice with GDC-0449 (Hh signalling antagonist) significantly reduced the number of Gli2 and CD31 (a commonly used capillarisation marker in vivo) double-positive cells in the liver (figure 9A,B). Similar results were observed in partial hepatectomised mice treated with cyclopamine (figure 9C). Hh inhibition thus prevents LSEC capillarisation in both chronic and acute liver injury.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Inhibition of Hedgehog (Hh) pathway prevents liver sinusoidal endothelial cell (LSEC) capillarisation in vivo. (A) Liver sections from dimethyl sulphoxide (DMSO) and GDC-0449-treated Mdr2−/− mice were double-stained for Gli2 (brown, Hh target gene) and CD31 (blue, capillarisation marker). Note that LSEC co-express Gli2 and CD31 (arrow). Scale bar: 10 μm. The number of Gli2/CD31 double-positive cells per field (B) was counted in five random fields per mouse, ***p<0.001, n=3. (C) Liver sections from vehicle and cyclopamine-treated partial hepatectomised (PHx) mice were stained for Gli2 and CD31, and the number of Gli2/CD31 double-positive cells was counted. **p<0.01, n=3. Cyc, cyclopamine.
Discussion
The current study demonstrates that Hh signalling is activated during LSEC capillarisation and plays a major role in controlling this process in cultured LSEC. Freshly isolated primary LSEC were shown to express both Hh ligands and receptor. We also found that LSEC expression of Hh ligands, signalling components and target genes increased significantly during culture-induced capillarisation. Manipulating Hh signalling via pharmacological, antibody-mediated, or genetic approaches altered the phenotype of cultured LSEC, proving that LSEC are Hh-responsive cells. Moreover, cultured LSEC required Hh signalling to orchestrate complex biological responses, such as the genesis of vascular tubes, because both cell migration and tube formation was blocked by Hh pathway inhibitors but enhanced by Hh pathway agonists. Indeed, capillarisation itself proved to be a Hh-dependent process because Hh pathway inhibitors partly reverted capillarised LSEC to their healthy differentiated phenotype, and completely prevented LSEC from becoming capillarised both in vitro and in vivo.
Several types of resident liver cells are capable of producing Hh ligands and/or responding to Hh ligands including hepatocytes,31 HSC,19 cholangiocytes22 and immune cells.46 Here we provide the first direct evidence that freshly isolated, primary LSEC produce Hh ligands (both Shh and Ihh). Capillarisation in vitro does not further increase LSEC expression of Hh ligands. Rather, the LSEC repress their production of the Hh ligand antagonist, Hhip, and this appears to be permissive for upregulating pathway activity because mRNA of Hh-regulated genes are induced. The importance of autocrine activation of Hh signalling is further supported by evidence that neutralising anti-Hh antibodies suppress Hh pathway activity in cultured LSEC. Both canonical47 and non-canonical (Smo independent)48 Hh signalling have been reported in other types of endothelial cells. Non-canonical pathways that are capable of activating Hh-regulated transcription factors may also be deployed as LSEC undergo capillarisation in vitro, because treating cultured LSEC with recombinant Shh fails to activate Hh signalling further (data not shown). It remains unclear if sinusoidal capillarisation in injured livers is also controlled mainly via autocrine mechanisms, or if Hh ligands that are released from neighbouring liver cells act in a paracrine fashion to enhance Hh signalling in LSEC. It is also conceivable that mechanical forces that occur during liver damage, such as changes in blood flow, might influence LSEC Hh signalling by altering primary cilia.49 ,50 The genetic approach that we used to knock down Smo expression in primary LSEC repressed Hh signalling, however, showing unequivocally that Hh activation in LSEC occurs at least partly through the canonical Hh pathway.
Inhibition of Hh signalling blocks VEGF-induced LSEC migration and tube formation. Hh signalling has been reported to interact with VEGF signalling by inducing VEGF51 and neuropilin52 (co-receptor for VEGF) expression. Our data here also suggest that Hh signalling may regulate VEGF receptor 1 and VEGF receptor 2 expression. This is very intriguing because VEGF signalling is an important regulator for angiogenesis, which is an essential process during development and many kinds of adult injury repair. Further study is needed to elucidate the link between these pathways.
The most intrigue finding of this study is that Hh signalling plays a key role in capillarisation. Although fenestrae did not reappear in cultured LSEC after blocking Hh signalling, the cells did regain a gene expression profile that is more typical of the fully differentiated LSEC in healthy livers. Moreover, loss of fenestrae during culture and two different types of liver injury were prevented by abrogating Hh signalling. There are several clinical implications for these novel findings. First, differentiated (non-capillarised) LSEC prevent HSC activation and cause activated, myofibroblastic HSC to revert to quiescence,53 while capillarised LSEC lose these actions.53 ,54 Our new data thus suggest that inhibiting Hh signalling in fibrotic livers may reduce liver fibrosis by promoting the restitution of fenestrated LSEC, which antagonise HSC activation, as well as by directly acting on the HSC themselves to prevent/reverse the myofibroblastic phenotype, as was suggested previously.19 Second, fenestrated LSEC have been proposed to play an important role in clearing chylomicron remnants.55 Because the accumulation of these remnants is believed to initiate atherosclerosis,56 evidence that Hh pathway inhibitors maintain LSEC fenestra suggests a novel approach to treat atherosclerosis. Finally, age-induced pseudo-capillarisation and injury-related capillarisation produce similar phenotypic changes in LSEC. Therefore, it will be intriguing to examine whether there is increased Hh pathway activity during ageing.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
-
Funding This work was supported by National Institutes of Health grant RO1 DK077794 (AMD).
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.