Article Text

Abstract
Background Acute pancreatitis has long been considered a disorder of pancreatic self-digestion, in which intracellular activation of digestive proteases induces tissue injury. Chemokines, released from damaged pancreatic cells then attract inflammatory cells, whose systemic action ultimately determines the disease severity. In the present work the opposite mechanism is investigated; that is, whether and how inflammatory cells can activate intracellular proteases.
Design Using mice either deficient for the CD18-α subunit of the membrane attack complex-1 (MAC-1) complex or tumour necrosis factor (TNF)α, as well as after depletion of leucocyte subpopulations, pancreatitis was induced by 7-hourly caerulein injections (50 μg/kg, intraperitoneally). Pancreatic acini were coincubated in vitro from wild-type and cathepsin-B-deficient animals with phorbol-12-myristate-13-acetate (PMA)-activated neutrophils and macrophages, caerulein or TNFα, and activities of trypsin, cathepsin-B and caspase-3 were measured, as well as necrosis using fluorogenic substrates. TNFα was inhibited with monospecific antibodies.
Results Deletion of CD18 prevented transmigration of leucocytes into the pancreas during pancreatitis, greatly reduced disease severity and abolished digestive protease activation. Depletion of neutrophils and macrophages equally reduced premature trypsinogen activation and disease severity. In vitro activated neutrophils and macrophages directly induced premature protease activation and cell death in pancreatic acini and stimulation of acini with TNFα induced caspase-3 activation and necrosis via a cathepsin-B and calcium-dependent mechanism. Neutralising antibodies against TNFα and genetic deletion of TNFα prevented leucocyte-induced trypsin activity and necrosis in isolated acini.
Conclusions The soluble inflammatory cell mediator TNFα directly induces premature protease activation and necrosis in pancreatic acinar cells. This activation depends on calcium and cathepsin-B activity. The findings from the present work further suggest that targeting TNFα, for which pharmaceutical agents are readily available, could be an effective treatment strategy that directly addresses the cellular causes of pancreatitis.
- TNFα
- premature protease activation
- acute pancreatitis
- CD18
- acute pancreatitis
- pancreas
- pancreatic enzymes
- trypsinogen activation peptide
- pancreatitis
- epithelial cell adhesion
- cell biology
- chronic pancreatitis
- tumour markers
- epigenetics
- liver cirrhosis
- cancer genetics
- molecular oncology
- pancreatic tumours
- pancreatic cancer
- pancreatic disease
- pancreatic pseudocyst
- pancreatic disorders
- cadherins
- acini
- adhesion molecules
Statistics from Altmetric.com
- TNFα
- premature protease activation
- acute pancreatitis
- CD18
- acute pancreatitis
- pancreas
- pancreatic enzymes
- trypsinogen activation peptide
- pancreatitis
- epithelial cell adhesion
- cell biology
- chronic pancreatitis
- tumour markers
- epigenetics
- liver cirrhosis
- cancer genetics
- molecular oncology
- pancreatic tumours
- pancreatic cancer
- pancreatic disease
- pancreatic pseudocyst
- pancreatic disorders
- cadherins
- acini
- adhesion molecules
Significance of this study
What is already known on this subject?
Premature intracellular zymogen activation is critical for the onset of pancreatitis and depends on cathepsin-B activity.
Previous concepts of pancreatitis have postulated that a pathological stimulus triggers premature and intra-acinar cell activation of digestive proteases, followed by acinar cell injury. Subsequently, chemokines and cytokines released from damaged acinar cells would attract infiltrating inflammatory cells whose role was largely confined to a systemic inflammatory response.
The systemic inflammatory response and subsequent organ failure determine the severity of pancreatitis.
What are the new findings?
Inflammatory cell infiltration and activation must now be counted among the initial events in the disease process and neutrophils and macrophages are the dominant drivers of premature protease activation and acinar cell necrosis.
Their action is mediated by the soluble inflammatory cell mediator tumour necrosis factor (TNF)α that can directly induce premature intracellular protease activation and necrosis in pancreatic acinar cells.
The activation of trypsinogen via TNFα depends on the activity of cathepsin-B and an intact calcium signal.
Significance of this study
How might it impact on clinical practice in the foreseeable future?
Establishing a causal treatment for pancreatitis is an urgent clinical need.
Targeting TNFα could be an effective treatment strategy directly addressing the cellular causes of pancreatitis.
As several pharmacological agents targeting TNFα are already available and in clinical use for other indications, randomised trials for pancreatitis should be considered in defined phases of pancreatitis.
Whether the benefit of TNFα inhibition on necrosis prevention outweighs the risk of greater susceptibility towards infection need to be determined.
Introduction
Acute pancreatitis, a fatal disease for 20% of severely affected patients, has long been considered a disorder of pancreatic self-digestion, in which premature and intracellular activation of digestive proteases induces tissue injury.1 ,2 Chemokines released from damaged pancreatic cells then attract inflammatory cells, whose systemic action ultimately determined the disease severity. Recent studies have found, however, that transmigration of leucocytes into the pancreas can occur before necrosis develops.3 We therefore investigated whether inflammatory cells can activate proteases rather than vice versa.
Employing an experimental model of pancreatitis in mice, either deficient for the CD18-α subunit of the membrane attack complex-1 (MAC-1) complex, or after selective depletion of neutrophils and macrophages, we found that preventing inflammatory cell transmigration into the pancreas greatly reduces disease severity as well as intracellular protease activation. In vitro incubation of pancreatic acini with phorbol-12-myristate-13-acetate (PMA)-activated neutrophils or macrophages directly induced intracellular trypsinogen activation and cell death. Protease activation and necrosis were both mediated via tumour necrosis factor (TNF)α in a cathepsin-B and calcium-dependent manner, and could be prevented using a monospecific anti-TNFα antibody.
These results challenge the notion of a premature protease activation/leucocyte activation sequence and indicate that inflammatory cells can, in turn, drive premature protease activation via secreted TNFα and in a cathepsin-B-dependent and calcium-dependent manner. Inhibiting TNFα would therefore directly address the cellular cause of pancreatitis.
Materials and methods
Materials
Caerulein was obtained from Sigma (Taufkirchen, Germany). Collagenase from Clostridium histolyticum (EC.3.4.24.3) was from SERVA (lot no. 14007, Heidelberg, Germany). Human myeloperoxidase was from Calbiochem (San Diego, California, USA). The substrate R110-(CBZ-Ile-Pro-Arg)2 was obtained from Invitrogen (Eugene, Oregon, USA) and AMC-(Suc-Ala2-Pro-Phe) from Bachem (Heidelberg, Germany). An Anyl amylase quantification kit was purchased from Roche (Grenzach-Whylen, Germany). The biologically active, phosphorylated cholecystokinin (CCK) octapeptide (Tyr(SO3H)27)-cholecystokinin fragment, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis (acetoxymethyl ester) (BAPTA-AM) and cycloheximide were obtained from Sigma (Eppelheim, Germany). All other chemicals were of highest purity and obtained either from Sigma-Aldrich (Eppelheim, Germany), Merck (Darmstadt, Germany), Amersham Pharmacia Biotech (Buckinghamshire, UK), or Bio-Rad (Hercules, California, USA).
Induction of pancreatitis in CD18-deficient, TNFα-deficient mice and depletion of neutrophils and macrophages
CD18-deficient mice were provided by Scharffetter-Kochanek et al.4 TNFα-deficient animals were obtained from Charles River (JaxMiceDatabase, Charles River Germany, strain: B6; 129S-Tnf tm1Gkl/J, stock number: 003008). Pancreatitis was induced in 14-week-old CD18-deficient mice and respective littermates by 7-hourly intraperitoneal caerulein injections (50 μg/kg bodyweight).3 ,5 All animals were starved overnight with access to water ad libitum.
Neutrophil depletion was induced by 100 μl anti-neutrophil serum (Accurate, New York, NY, USA) injected intraperitoneally 24 h before induction of pancreatitis in C57BL6 mice.
The depletion of macrophages/monocytes was performed by intraperitoneal injection of 100 μl clodronate-containing liposomes (VU Medisch Centrum, Amsterdam, The Netherlands) 3 days and 1 day before induction of pancreatitis.6 Intraperitoneal injection of phosphate-buffered saline (PBS) containing liposomes served as a control. All animals used in this study were maintained according to institutional guidelines and animal facility protocols. All experiments including induction of pancreatitis were performed after prior approval by the institutional animal care committee.
Preparation of serum and tissue samples
Mice were killed 0 h, 8 h and 24 h after the first injection of caerulein. After exsanguination under ether anaesthesia the pancreas was transferred to OCT Compound (Tissue Tek, Sakura Finetek, Torrance, California, USA) for cryosections or fixed in 5% paraformaldehyde for paraffin embedding. The main part of the pancreas was frozen in liquid nitrogen and stored at −80°C for later analysis.3 ,7 Serum was sampled and stored at −20°C. Tissue samples for measurement of pancreatic enzyme activity were homogenised on ice in a buffer containing 100 mM Tris, 5 mM CaCl2 pH 8.0. The supernatant was separated and used for fluorometric enzyme measurement.
Biochemical assays
Amylase and lipase activities were measured by photometric assays (Roche, Grenzach-Whylen) and standardised using purified enzymes (Sigma). The cytokines monocyte chemotactic protein 1 (MCP-1) and TNFα in serum were measured by fluorescence-activated cell sorting (FACS) analysis with the CBA mouse inflammation kit according to the manufacturer's instructions (Becton Dickinson, Heidelberg, Germany). Trypsin activity was measured by fluorometric enzyme kinetic over 1 h at 37°C using rhodamine-110-CBZ-Ile-Pro-Arg (Invitrogen, Karlsruhe, Germany) as substrate. The trypsin activity of samples was corrected for protein content (Bradford assay).
Myeloperoxidase (MPO) activity measurement was performed as previously described.8 Briefly, pancreatic tissue was homogenised on ice in 20 mM potassium phosphate buffer (pH 7.4) and centrifuged. The pellet was resuspended in 50 mM potassium phosphate buffer (pH 6.0) containing 0.5% cetyltrimethylammoniumbromide. The suspension was freeze-thawed in cycles, sonicated and centrifuged at 20,000 g. MPO activity was assayed in 50 mM potassium phosphate buffer (pH 6) containing 0.53 mM O-dianisidine and 0.15 mM H2O2. The initial increase in absorbance was measured at room temperature with a Spectramax Spectrophotometer. The results are expressed in units of MPO activity. Bars indicate mean values in U MPO activity per g pancreatic protein (± SEM) from five or more animals per timepoint.
Preparation of acinar cells and splenocytes
Leucocytes were isolated from murine spleen of CD18+/+, CD18−/− and C57Bl6 control mice by using cell filters (70 μm). After lysis of erythrocytes with 155 mM NH4Cl, 10 mM KHCO3 and 0.1 mM EDTA, cells were counted and transferred to sterile PBS. Macrophages and neutrophils were isolated from total splenocytes using MACS cell separation kits according to the manufacturers instructions (Miltenyi Biotech, Bergisch-Gladbach, Germany) with respective antibodies, Ly6 being specific for neutrophils and CD11c being specific for macrophages. Purity of cells was >90% as confirmed by FACS cell sorting. Cells were washed three times after activation with 100 nM PMA for 15 min and then transferred for coincubation with pancreatic acinar cells.
Acinar cells were prepared from CD 18−/−, CD18+/+,4 C57BL6 and cathepsin-B−/−9 mice. Acinar cells were prepared by collagenase digestion, maintained and stimulated in Dulbecco's modified Eagle medium containing 2% bovine serum albumin (BSA) and 10 mM 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (HEPES). Acinar cells were stimulated with 0.001 mM CCK and, as shown in supplementary figure 1, pretreated with 300 μM cyclohexemide (CHX) to inhibit protein synthesis.10 Calcium signalling was blocked by the calcium chelator BAPTA-AM at a concentration of 10 μM. Coincubation of freshly isolated acinar cells with a crude splenocyte extract, isolated macrophages or neutrophils was performed after stimulation with CCK, and additionally with an anti-TNFα antibody 100 μg/ml (infliximab, Essex Pharma, Munich, Germany). Acinar cells were stimulated with murine recombinant TNFα (10 ng/ml) and interleukin (IL)-1β (10 ng/ml) (Sigma, Taufkirchen) either with or without CCK stimulation.7 Trypsin activity was measured by rhodamine 110 substrates as previously described11 at a concentration of 10 μM, cathepsin-B was measured using 20 μM AMC-Arg2 and caspase-3 activity by PhiPhiLux (Calbiochem, Darmstadt) at a final concentration of 350 μM. Acinar cell necrosis was quantitated by propidium iodine exclusion.5 ,11
Western blot analysis
Tissue samples for western blot experiments were homogenised on ice in lysis buffer containing 25 mM HEPES (pH 7.5), 75 mM NaCl, 0.5% Triton X-100, 5% glycerol, 1 mM EDTA in the presence of different protease inhibitors (10 mM NaF, 5 mM Na4P2O7, 1 mM PMFS and 1 μg/ml aprotinine). Protein content was determined by the Bradford assay. In all, 100 μg samples of total protein were loaded on 12.5% polyacrylamide gels and transferred to nitrocellulose membranes for immunoblotting as previously described.3 CD18 antibody (Abcam rat monoclonal clone C71/16) was used at a dilution of 1:1000 and anti-β-actin used as loading control.
Morphology and morphometry
At selected timepoints of pancreatitis, pancreas tissue samples were collected from CD18−/− and CD18+/+ mice, TNFα-depleted animals, and neutrophil-depleted or macrophage-depleted animals and their respective controls. Pancreatic tissue was immediately fixed for paraffin histology and cryosections. Paraffin sections were used for haematoxylin and eosin staining as previously described.8 Macrophages were detected by immunohistochemical staining with F4/80 antibody (AbD serotec rat monoclonal CI:A3-1), diluted 1:25 in 1% Aureon-BSA (Aureon Biosystems, Vienna, Austria); CD18 antibody was diluted 1:100.
Statistical analysis
All data are expressed as means ± SEM from at least five animals in each group. Statistical analyses of the different mouse groups were made by GraphPad Prism and Sigma-Plot using the Student t test for independent samples. Differences were considered significant at a level of p<0.05.
Results
Impaired leucocyte transmigration in CD18-deficient mice abolishes protease activation and ameliorates acute pancreatitis
Pancreatitis causes an inflammatory response accompanied by increased transmigration of leucocytes into the pancreas.3 Here, we examined the effect of leucocyte transmigration during pancreatitis in CD18+/+ and CD18−/− mice.4 ,12 CD18 is an essential component of the β2-integrin family of leucocyte adhesion molecules. These heterodimeric molecules consist of a common β-subunit designated as CD18, which is non-covalently bound to the α-subunit CD11a (LFA-1), CD11b (Mac-1), CD11c (gp150/95) or CD11d. CD18 deficiency in mice results in virtually complete recruitment failure of neutrophils to sites of toxic or allergic dermatitis as well as skin wounds.13
In experimental pancreatitis of CD18−/− mice the transmigration of leucocytes was nearly abolished at 8 h (figure 1A–C), and associated with a reduction in local tissue damage (figure 1D) and in intrapancreatic trypsin activity (by >70%) (figure 1E). Interestingly, 24 h after the onset of pancreatitis leucocytes had apparently entered the pancreas in a CD18-independent manner (figure 1B,C)—possibly via injured endothelia—and this was paralleled by an equivalent rise in tissue damage and protease activation. While these data establish a link between the presence of inflammatory cells in the pancreas, premature protease activation and acinar cell injury, they can not prove a direct cause and effect relationship. We therefore coincubated living pancreatic acini with PMA-activated mouse splenocytes in vitro and quantitated intracellular trypsin activity. Splenocytes by themselves were able to induce intracellular protease activation in pancreatic acinar cells to an extent observed after supramaximal CCK stimulation alone. This protease activation was associated with an equally pronounced and thus significant increase in necrosis as induced by CCK stimulation alone of up to 15±2% of acinar cells (necrosis not shown). These experiments (figure 1F) show a direct induction of intracellular trypsinogen activation in living acinar cells by splenocytes and prompted us to distinguish between subpopulations of inflammatory cells.
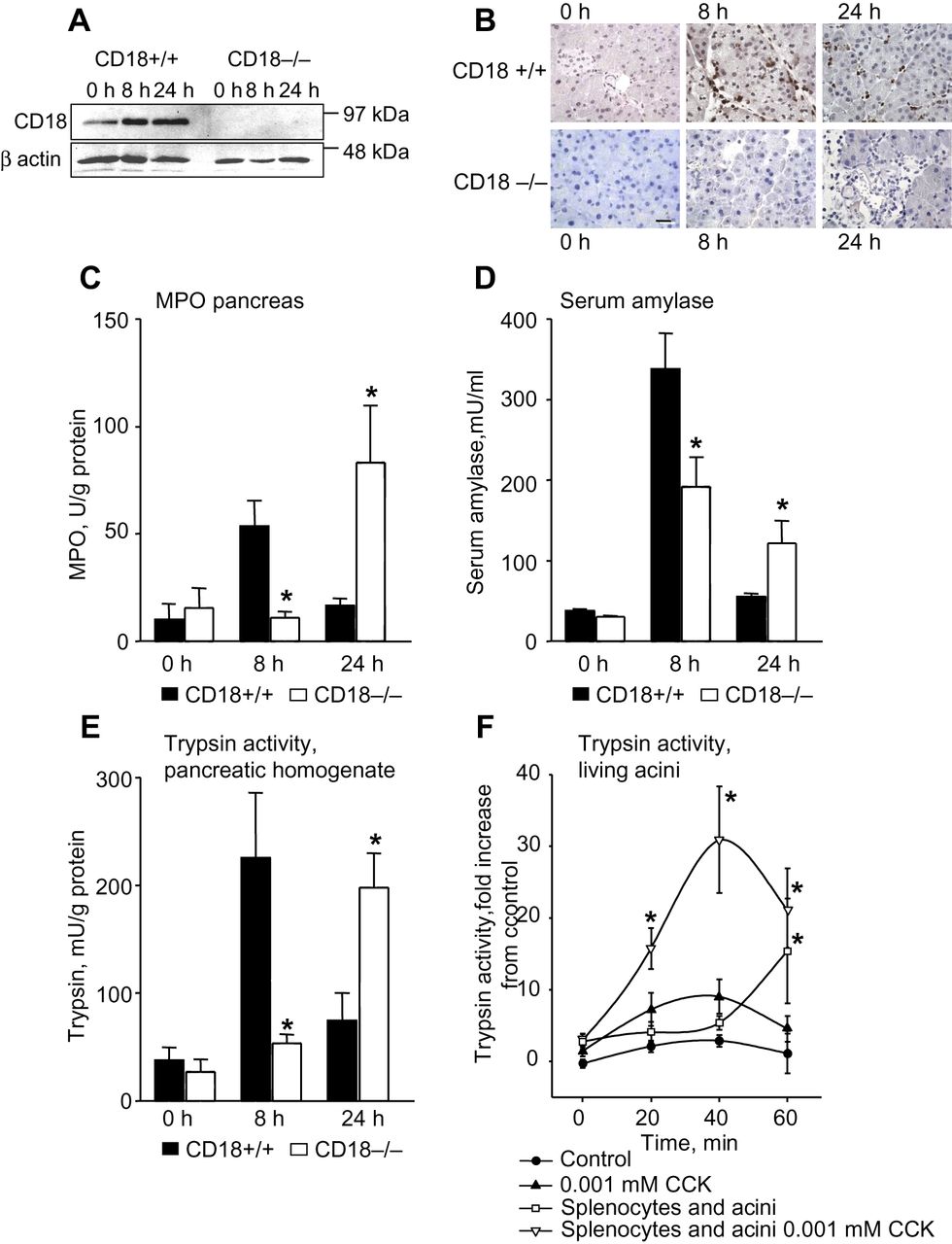
Experimental pancreatitis in CD18-deleted mice. During the course of pancreatitis no CD18 expression was detected in tissue (spleen) of CD18 knockout animals (A). In wild-type mice immunolabelling for CD18 indicated a peak of leucocyte infiltration in the pancreas after 8 h of pancreatitis whereas in CD18−/− animals leucocyte infiltration was only detected after 24 h (B). This corresponded to a peak of myeloperoxidase (MPO) activity after 8 h in wild-type animals and one at 24 h in CD18−/− mice (C). Severity, as indicated by serum amylase activity, was also greatest at 8 h of pancreatitis in wild-type and at 24 h in CD18−/− mice (D). Intrapancreatic trypsin activity (E) during experimental pancreatitis completely paralleled MPO activity (C). In vitro coincubation of phorbol-12-myristate-13-acetate (PMA)-activated splenocytes with freshly isolated pancreatic acini resulted in a fivefold increase in intracellular trypsinogen activation over incubation with supramaximal cholecystokinin alone (F). More than five animals were used for each experiment and all experiments were performed as triplicates. All experiments were performed independently on three or more occasions. Asterisks indicate significant differences with p<0.05.
Depletion of neutrophils and macrophages ameliorates caerulein induced pancreatitis and reduces protease activation
One option for reducing neutrophils in the systemic circulation, rather than blocking their transmigration into damaged tissue, is the use of anti-neutrophil serum.14–16 This greatly reduced morphological damage during pancreatitis (figure 2A), almost abolished the presence of neutrophils in the pancreas (figure 2B) and reduced disease severity (figure 2C) and intrapancreatic trypsin activity (figure 2D). The latter reduction was, however, less than that found in CD18−/− animals, suggesting that neutrophils are not the only inflammatory cells involved in premature protease activation.

Course of caerulein-induced pancreatitis following depletion of neutrophils. Haematoxylin and eosin staining of the pancreas during pancreatitis demonstrates reduced pancreatic injury and less extensive inflammatory infiltrates in neutrophil-depleted animals (A). This treatment with anti-neutrophil serum (AN-serum) reduced myeloperoxidase (MPO) activity to control levels in pancreatitis (B) and serum amylase activity (C), and trypsinogen activation (D) by up to 50%. Asterisks indicate significant differences with p<0.05.
As other investigators had proposed a role for macrophages in pancreatitis17 ,18 and we had found MCP-1 to be elevated as early as 60 min after the onset of pancreatitis (not shown), we used intraperitoneal clodronate liposomes to deplete monocytes/macrophages in vivo before the induction of pancreatitis. Unlike in the normal liver, macrophages are barely detectable in the pancreas of control animals but their numbers increase dramatically during pancreatitis (figure 3A). This rise in the pancreas and their presence in the liver were completely prevented by intraperitoneal clodronate (100 μl). While this does not affect the neutrophils in the pancreas (as indicated by increased pancreatic MPO, figure 3B), it reduced tissue damage (figure 2C) and trypsinogen activation (figure 3D) to a similar extent as the depletion of neutrophils with antiserum.

Course of caerulein-induced pancreatitis following depletion of macrophages/monocytes. Macrophages/monocytes were depleted with clodronate liposomes before the induction of pancreatitis (A). This treatment completely eliminated F4/80-positive macrophages/monocytes from the liver and prevented their appearance in the pancreas, during the time course of pancreatitis (A). Clodronate depletion did not affect the rise in myeloperoxidase (MPO) in the pancreas (B) but reduced the rise in serum lipase (C) and intrapancreatic trpysinogen activation (D). The experimental group as well as the control group consisted of five animals each and all measurements were performed in triplicates. Asterisks indicate significant differences with p<0.05.
Macrophages and neutrophils can directly evoke intracellular protease activation and cell necrosis in pancreatic acinar cells
Having established this in vivo relationship between the transmigration of neutrophils and monocytes into the pancreas and the evolution of protease activation and pancreatic damage, we tested whether this was a direct and causal association by incubating either MACS-bead-sorted neutrophils or macrophages with isolated acinar cells in vitro. The coincubation with neutrophils (figure 4A,B) as well as with macrophages (figure 4C,D) induced trypsinogen activation inside isolated acinar cells. Their presence also greatly increased the extent of protease activation and acinar cell necrosis induced by coincubation with supraphysiological secretagogue concentrations. For neutrophils we could even demonstrate the induction of necrosis in the absence of secretagogue (figure 4B).
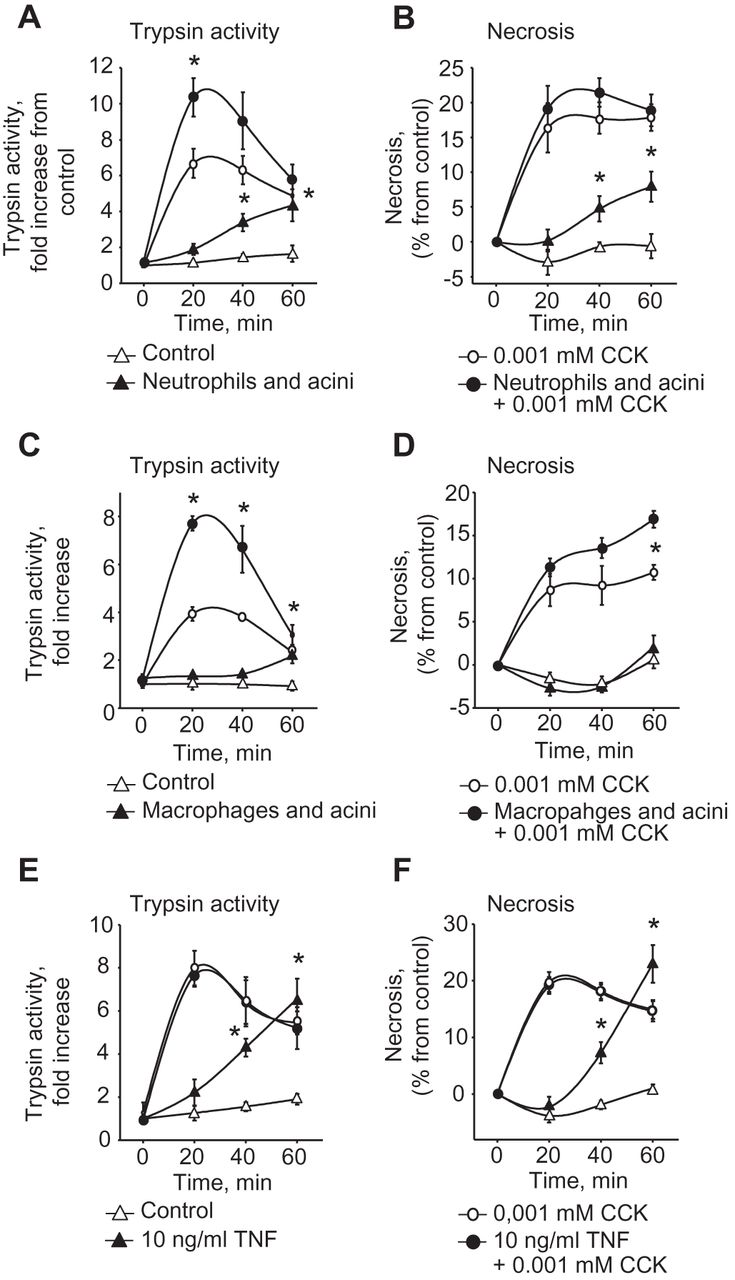
Direct effects of neutrophils, macrophages and tumour necrosis factor (TNF)α on pancreatic acinar cells. Isolated splenocytes, neutrophils and macrophages were coincubated with freshly isolated pancreatic acini. Isolated neutrophils combined with caerulein doubled the rate of trypsinogen activation in acini over the extent achieved with supramaximal stimulation alone (A), and induced trypsinogen activation and acinar cell necrosis (B) in the absence of cholecystokinin (CCK) stimulation. Isolated macrophages increased the rate of trypsinogen activation (C) and acinar cell necrosis (D) over that found after supramaximal CCK stimulation alone. Acini were incubated with tumour necrosis factor (TNF)α (10 ng/ml). TNFα induced trypsinogen activation as well as necrosis (F). Acinar cells and leucocytes subpopulations were prepared for at least five different assays and measurements were performed in triplicates.
These experiments raise the question whether a soluble factor released from inflammatory cells mediates this dramatic effect on protease activation in acinar cells and affects their viability. Two candidates that would both be secreted by neutrophils and monocytes and which have previously been implicated in the systemic complications of pancreatitis are TNFα19 ,20 and IL-1β.21 We used both for in vitro studies and found that IL-1β does not affect premature protease activation or induce acinar cell necrosis (data not shown) whereas the presence of TNFα alone was entirely sufficient to induce both (figure 4E,F). This suggests that activation of the innate immune system can, in itself, be sufficient to induce pancreatitis, and that external events, such as pancreatic duct obstruction by an impacted gallstone22 or pathological hormone or ethanol stimulation,1 are not an indispensable prerequisite.
TNFα mediates trypsin activity and necrosis via cathepsin-B and this effect is blocked by anti-TNFα antibody
The fact that neutrophils and monocytes induce protease activation and cell injury via TNFα raises the question how this effect is mediated and whether it can be antagonised. We found that TNFα increases caspase-3 and cathepsin-B activity in living acini (figure 5A,B). Previous studies from our laboratory had shown that lysosomal cathepsin-B is critical for intrapancreatic trypsin activation.9 We repeated these experiments in acini from cathepsin-B-knockout animals and found that the TNFα effect on pancreatic protease activation and necrosis disappeared (figure 5C,D). It is well established that trypsin activity is induced by a sustained intracellular calcium peak following supramaximal CCK stimulation. Incubation of pancreatic acinar cells with the AM ester of the calcium chelator BAPTA, a compound that has been shown to bind intracellular calcium with high affinity in pancreatic acinar cells, abolished intracellular trypsinogen activation in response to CCK and in response to TNFα stimulation (figure 5E). We further excluded that newly synthesised cathepsin-B rather than activation of preformed cathepsin-B is responsible for intracellular trypsinogen activation by incubating pancreatic acini for 60 min with the protein synthesis inhibitor cyclohexemide (300 μM) (figure 5F,G), which did not affect cathepsin-B activity or the processing of preformed cathepsin-B (figure 5G). The action of TNFα on acinar cell trypsinogen activation and injury is thus completely dependent on the presence of cathepsin-B and an intact intracellular calcium signal. This disease mechanism would be of even greater relevance if it could be directly addressed by a targeted therapeutic intervention. We therefore used infliximab, a specific anti-TNFα antibody, to block TNFα in vitro and this resulted in reduced leucocyte-mediated trypsinogen activation, reduced cathepsin-B activity and reduced acinar cell necrosis (figure 6A–C). This confirms the direct link between leucocyte-mediated cell damage and its action via cathepsin-B-mediated protease activation, and also suggests that inhibition of TNFα would directly address a cellular cause of pancreatitis. To study whether a deletion of TNFα would ameliorate the course of caerulein induced pancreatitis we evaluated local pancreatic damage, myeloperoxidase and trypsin activity in the pancreas of TNFα-knockout mice. TNFα knockout resulted in less pronounced inflammatory infiltration and less necrosis on haematoxylin and eosin staining (figure 6D). Moreover, MPO activity was significantly reduced, as was intrapancreatic trypsin activity after 8 h of caerulein pancreatitis (figure 6E,F). TNFα inhibitors could now be a feasible option for treating patients with pancreatitis in defined phases of the disease.

Effect of tumour necrosis factor (TNF)α on cathepsin-B-mediated trypsin activation and necrosis. TNFα stimulation of pancreatic acinar cells induces caspase-3 activation (A) and intracellular cathepsin-B activity (B). In cathepsin-B-depleted animals the effect of TNFα on caspase-3 activation (C) and on acinar cell necrosis (D) was abolished. A blocked intracellular calcium signal by the 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis (acetoxymethyl ester) (BAPTA-AM) abolished intracellular trypsinogen activation upon supramaximal cholecystokinin (CCK) and TNFα stimulation in isolated pancreatic acinar cells from C57BL6 mice (E). Acini pretreated with cycloheximide (CHX) 300 μM and stimulated with 10−7 mM CCK showed no difference in cathepsin-B activity compared to control (F). In western blot analysis (of two independent experiments) from CCK stimulated acini the 42 kDa band depicted as pro-cathepsin-B disappeared indicating cathepsin-B processing. We did not observe a change in the processing of pro-cathepsin-B to mature cathepsin-B in the presence of CHX indicating that CCK acts on preformed cathepsin-B (G). Acinar cells were prepared for at least three different assays and measurements were performed in triplicates. Asterisks indicate significant differences with p<0.05.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of tumour necrosis factor (TNF)α inhibition by monospecific antibody on cathepsin-B-mediated trypsin activation and necrosis. Adding an anti-TNFα antibody (infliximab 100 μg/ml) to acini coincubated with activated splenocytes significantly reduced trypsinogen activation (A), acinar cell necrosis (B) and cathepsin-B activity (C). We noted a reduced inflammatory infiltrate and less necrosis in TNFα knockout animals on haematoxylin and eosin staining (D), which was paralleled by reduced pancreatic myeloperoxidase (MPO) activity (E). In line with the in vitro data we found a significantly reduced trypsin activity in pancreatic homogenates of TNFα knockout animals. All results were generated from five animals in each group and all measurements were performed in triplicates. Asterisks indicate significant differences with p<0.05.
Discussion
Acute pancreatitis is a common abdominal disorder and patients affected by its severe variety have a high mortality rate.23 For more than a century self-digestion of the pancreas by its own proteolytic enzymes has been regarded as the principal cause of the disease.2 ,24 It is generally accepted that pancreatitis begins in acinar cells,25 that cell injury involves intracellular trypsinogen activation in a cathepsin-B-dependent and calcium-dependent manner,9 ,10 and that mutations in the cationic trypsinogen gene (PRSS1) represent potent risk factors for developing pancreatitis in man.26 Protease-induced cellular injury causes the release of chemokines, which in turn attract inflammatory cells. Their systemic actions determine the ultimate disease severity.14 ,17 Recent studies have shown that inflammatory cells and their cytokines cause the systemic complications of pancreatitis, such as lung injury and shock, and are also involved in the local damage within the pancreas.15 ,16 ,21 ,27 Moreover, the transmigration of inflammatory cells into the pancreas appears to occur within minutes, rather than hours, of the disease onset and thus at timepoints before acinar cell necrosis has evolved.3 ,28 ,29 This prompted us to investigate whether and how intrapancreatic protease activation and cell injury can actually be induced by infiltrating inflammatory cells rather than being the cause of inflammation.
The CD18-deficient mouse represents an ideal model to test the contribution of leucocytes to protease activation and disease severity of pancreatitis. Cells of the innate immune system, whose extravasation and transmigration depend on CD18, would be the first cells to reach the pancreas in the course of pancreatitis.30–33 In our study transmigration of leucocytes was abolished by CD18 deletion and this was associated with a more than 70% reduction in protease activation. At 24 h after the onset of pancreatitis the leucocytes of CD18-deficient mice began to transmigrate independently of CD18 into the pancreas, as reflected by an increase in MPO and leucocyte-specific labelling. This was paralleled by an equally prominent increase in protease activation indicating that transmigrating leucocytes mediate the severity of pancreatitis and protease activation. This direct link between leucocyte infiltration and protease activation in the pancreas was confirmed by experiments in which the coincubation of pancreatic acini with inflammatory cells isolated from spleen (splenocytes) directly induced premature intracellular zymogen activation and necrosis in the former. These data prompted us to further investigate the role of different leucocyte subpopulations on premature intracellular zymogen activation and cell damage.
A hallmark of acute pancreatitis is the accumulation of neutrophils in the pancreas and their depletion was shown to attenuate the severity of pancreatitis and associated lung injury.3 ,14–16 ,34 Little, however, is known about the direct effects of neutrophils on acinar cells and the mechanisms mediating cell death in the latter. Elegant in vivo studies have previously shown that the absence of neutrophils during pancreatitis reduces the extent of tissue damage in the pancreas and that a mechanism mediating cell injury is the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase.16 Our experiments using antibody depletion of neutrophils confirm this effect and suggest that a direct action of neutrophils on intra-acinar cell protease activation contributes to this process. However, the effect of CD18 deletion in our study on either necrosis or protease activation was found to be much greater than that of neutrophil depletion alone, suggesting that other leucocyte subpopulations must also be involved.
Several lines of evidence from previous studies18 ,29 as well as our observation that serum levels of MCP-1 increase as early as 1 h after the onset of pancreatitis suggest that macrophages play a very early role in acute pancreatitis. The data presented here indicate that the in vivo depletion of macrophages reduces pancreatitis severity and intrapancreatic protease activation, and also that a direct interaction between isolated macrophages and acinar cells in vitro involves an induction of protease activation and necrosis that is comparable to that induced by neutrophils. The question remains by what mechanism neutrophils and macrophages induce intracellular protease activation.
Macrophages are a major source of TNFα and IL-1β, cytokines, whose serum levels parallel disease severity of acute pancreatitis and both of which markedly increase during the early disease course.35 ,36 We therefore sought to determine whether TNFα or IL-1β would trigger premature intracellular zymogen activation directly and thus participate in the signalling pathway leading to necrosis. We found that TNFα, but not IL-1β, induced premature intracellular protease activation and did so to the same extent as supramaximal CCK stimulation alone. TNFα stimulation resulted in a twofold increase in caspase activity over control after 1 h, while we detected a 15% propidium iodine uptake in the cells in line with previous reports outlining the role of TNFα in necrosis formation.37 Gukovskaya and coworkers found a rate of necrosis in rat pancreatic acinar cells of 8% after 6 h of TNFα stimulation.19 ,20 This difference might be due to the sensitivity of the method and the different species employed (rats vs mice). Lactate dehydrogenase release is known to be less sensitive than uptake of the fluorogenic cell dye propidium iodine. Moreover rat pancreatic acinar cells are much more resistant to necrosis compared to mice acini. With regard to the rate of apoptosis we found a twofold increase of caspase-3 activity after 60 min, and in line with this finding Gukovskaya and coworkers reported 13% apoptotic cells after a much longer incubation period of 6 h.
The absence of an IL-1β effect on acini was surprising because the pancreas-specific transgenic overexpression of IL-1β in mice had been shown to lead to chronic pancreatitis.21 Since our data rule out a direct effect of IL-1β on acinar cells its role in tissue damage must be indirect and probably involves the activation of neutrophils and macrophages.
Unlike for IL-1β, a direct effect of TNFα on acinar cells could clearly be shown. This is less surprising because the presence of TNFα receptors on pancreatic exocrine cells had already been demonstrated and they were mostly implicated in the regulation of acinar cell survival and apoptosis.19 ,20 ,38 During apoptosis TNFα is thought to be involved in two distinct pathways, one involving the activation of caspase-3, a process we could confirm in our studies, and one involving the breakdown of mitochondrial function.39–41 Both kinds of apoptosis, however, have previously been thought to reduce, rather than increase, the severity of pancreatitis because, unlike necrosis, they are not followed by a systemic inflammatory response.42 It is in this systemic inflammatory response where a role for TNFα, released by macrophages and neutrophils, has been established, and mainly involves the activation of nuclear factor (NF)κB.43–45
Our study provides evidence for quite a different role of TNFα in pancreatitis. In experiments using isolated acini, neutrophils as well as macrophages directly induced intra-acinar cell protease activation and necrosis and TNFα was identified as their common mediator. TNFα alone induced trypsin activation and necrosis to an extent comparable to supramaximal concentrations of caerulein. In further experiments we were able to show that this effect is entirely dependent on an intact intracellular calcium signal. However, TNFα-induced trypsinogen activation and necrosis were absent in acini from cathepsin-B-deficient animals, which indicates that this process is dependent on calcium and cathepsin-B. Activation of cathepsin-B was independent of de novo protein synthesis. We hypothesise that TNFα stimulation leads to lysosomal permeabilisation via activation of caspase-8 and truncated BH3 interacting domain death agonist (tBID) phosphorylation and thus the release of active cathepsin-B into the cytoplasm.40 ,46 The activation of trypsinogen by cathepsin-B after TNFα stimulation would most likely occur in cytoplasmic vesicles, that could also undergo permeabilisation. Whether and how this event is regulated is the topic of another ongoing project.
The capacity of neutrophils and macrophages to induce protease activation and necrosis in isolated acinar cells was abolished by treatment with monospecific anti-TNFα antibodies and in a TNFα-knockout model. This observation explains the beneficial effect of deletion of TNFα receptor 1 on the severity of experimental pancreatitis,47 a process that could not be explained by effects on TNFα-mediated apoptosis alone. Furthermore, late mortality in severe acute pancreatitis can be predicted by sustained levels of increased TNFα and a reduction of human leukocyte antigen (HLA)-DR expression on monocytes, which indicates an ongoing compensatory anti-inflammatory response syndrome (CARS).48 Genetic polymorphisms in the TNFα gene are associated with an increase in mortality in severe acute pancreatitis.49 However, data from sepsis trials on the use of TNFα blockage are somewhat contradictory50 suggesting that the timepoint of blockage is essential and TNFα is only beneficial in ongoing CARS. To ensure a beneficial effect it might therefore be necessary to establish CARS in a clinically relevant cohort of patients with severe acute pancreatitis before anti-TNFα treatment can be tested. However, the cellular infiltrate found in resection specimens from patients with chronic pancreatitis is composed of TNFα producing cells, such as macrophages, neutrophils, T cells and stellate cells.51 Our finding that TNFα directly induces protease activation in pancreatic acinar cells suggests that pharmacological inhibition of TNFα signalling might be as effective in treating chronic pancreatitis as it is in Crohn's disease or rheumatoid arthritis. Whether the benefits of TNFα inhibition on premature protease activation outweigh the risk of increasing the susceptibility towards infection needs to be explored before clinical trials on pancreatitis can be designed.
Our results have a number of consequences: previous concepts of pancreatitis have postulated that a pathological stimulus, be it pancreatic duct obstruction by an impacted gallstone or toxic exposure to ethanol or its metabolites, triggers premature and intra-acinar cell activation of digestive proteases and then acinar cell injury. Subsequently the chemokines and cytokines released from damaged acinar cells would attract the infiltration of inflammatory cells whose role was largely confined to the systemic inflammatory response. The new concept would suggest that inflammatory cell infiltration and activation must be counted among the initial events of the disease process and that neutrophils and macrophages are the dominant drivers of premature protease activation and acinar cell necrosis, that they mediate this effect via TNFα and that it completely depends on the activity of cathepsin-B. This improves our understanding of the pathophysiology of pancreatitis and offers new therapeutic options. As several pharmacological agents targeting TNFα are already available and in clinical use for other indications randomised trials for pancreatitis should be considered. Such agents could be an effective treatment strategy that directly addresses the cellular causes of pancreatitis.
Acknowledgments
We would like to thank Kathrin Gladrow for her excellent technical assistance.
References
Footnotes
MML and JM contributed equally to the study.
Funding Supported by the Alfried-Krupp-von-Bohlen-und-Hahlbach-Foundation (Graduate Schools Tumour Biology and Free Radical Biology), the Deutsche Krebshilfe/Dr Mildred-Scheel-Stiftung (109102), the Deutsche Forschungsgemeinschaft (DFG GRK840-E3/E4, MA 4115/1-2/3, NI 1297/1-1), the Federal Ministry of Education and Research (BMBF GANI-MED 03152061A and BMBF 0314107) and the European Union (EU-FP-7: EPC-TM and EU-FP7-REGPOT-2010-1).
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.