Article Text

Abstract
Objective Cholestasis is associated with increased liver injury and morbidity after partial hepatectomy (PH), yet bile acids (BAs) are emerging as important mediators of liver regeneration. Fibroblast growth factor 15 (Fgf15, human FGF19) is a BA-induced ileum-derived enterokine that governs BA metabolism. We evaluated the relevance of Fgf15 in the preservation of BA homeostasis after PH and its potential role in the regenerative process.
Design Liver regeneration after PH was studied in Fgf15 −/− and Fgf15 +/+ mice. The effects of the BA sequestrant cholestyramine and adenovirally delivered Fgf15 were examined in this model. The role of Fgf15 in BA-induced liver growth was tested in Fgf15 −/− mice upon cholic acid (CA) feeding. The direct mitogenic effect of Fgf15 was evaluated in cultured mouse hepatocytes and cholangiocytes.
Results Fgf15 −/− mice showed marked liver injury and mortality after PH accompanied by persistently elevated intrahepatic BA levels. Cholestyramine feeding and adenovirally delivered Fgf15 reduced BA levels and significantly prevented this lethal outcome. Fgf15 also reduced mortality after extensive hepatectomy in Fgf15+/+ animals. Liver growth elicited by CA feeding was significantly diminished in Fgf15 −/− mice. Proliferation of hepatocytes and cholangiocytes was also noticeably reduced in CA-fed Fgf15 −/− mice. Fgf15 induced intracellular signalling and proliferation of cultured hepatocytes and cholangiocytes.
Conclusions Fgf15 is necessary to maintain BA homeostasis and prevent liver injury during liver regeneration. Moreover, Fgf15 is an essential mediator of the liver growth-promoting effects of BA. Preoperative administration of this enterokine to patients undergoing liver resection might be useful to reduce damage and foster regeneration.
- Growth Factors
- Liver Regeneration
- Bile Acid
- Acute Liver Failure
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
-
Bile acids (BAs) have been recently recognised as key signalling molecules in the regulation of liver regeneration.
-
After liver resection, homeostasis of BAs needs to be tightly controlled to prevent cholestasis while allowing proper BA signalling in the regenerative process.
-
Portal blood is known to contain essential mediators of liver regeneration. Fibroblast growth factor 15 (Fgf15 in rodents, FGF19 in humans) is a BA-induced ileum-derived enterokine that governs BA homeostasis.
What are the new findings?
-
Fgf15 is necessary to modulate BA levels during liver regeneration after partial hepatectomy (PH). Fgf15 −/− mice with PH display very high mortality due to toxic intrahepatic BA concentrations.
-
Delivery of Fgf15 to the hepatic parenchyma reduces intrahepatic BA levels and improves survival after liver resection in Fgf15 −/− mice, and increases survival after extensive liver resection in wild type mice.
-
Fgf15 is a key mediator of the liver growth-promoting effects of BA. Fgf15 is a direct mitogen for hepatocytes and biliary epithelial cells.
How might it impact on clinical practice in the foreseeable future?
-
New strategies are needed to improve the function and regeneration of remnant livers after resection and liver grafts during living donor transplantation.
-
Preoperative administration of FGF19, the human orthologue of Fgf15, to patients may prevent cholestasis and liver dysfunction after major resection or transplantation.
-
Administration of this enterokine may improve the regenerative capacity of the liver by enhancing the proliferation of parenchymal and biliary epithelial cells.
Introduction
Liver resection is a relatively safe procedure carried out for the treatment of benign and malignant hepatic and metastatic tumours and in living donor liver transplantation. The feasibility of this intervention relies to a great extent on the regeneration of the remaining parenchyma. Failure to mount a successful regenerative response often has fatal consequences unless a transplant can be performed.1–4 The liver has a tremendous regenerative potential, unmatched by any other tissue in the adult organism.5 Over the past decades valuable information on the cellular and molecular mechanisms underlying this process has been gathered in experimental models of hepatic regeneration after partial hepatectomy (PH).5–8 In addition to the primary relevance of this knowledge, there is also a clear translational interest in the identification of the key molecules that orchestrate the complex regenerative response. Certainly, the availability of pharmacological tools allowing the modulation of liver regeneration might improve the prognosis of many patients after liver resection.3
Hepatic regeneration entails the activation of multiple regulatory pathways. These pathways need to be very well coordinated to allow proper restoration of the tissue while maintaining vital liver functions. The nature of the molecules and signals that participate in the regenerative response is diverse, and three major types of pathways have been identified: the cytokine, growth factor and metabolic networks.6 Endocrine crosstalk of the liver with other tissues is known to be important for the regenerative process and such crosstalk may involve molecules within the three mentioned networks.5 ,6 ,9 Early observations in canine models already suggest the importance of portal blood borne substances, leading to the identification of the key role played by insulin in liver regeneration.8 More recently, fluctuations in the circulating and intrahepatic levels of bile acids (BAs) have been observed upon PH. BA signalling has been implicated in the initiation of the hepatoproliferative response in rodents10–13 and in liver growth after portal vein embolisation in humans.14 This newly realised effect of BA involves the activation of the farnesoid X receptor (FXR), the primary sensor of BA, as liver regeneration is defective in FXR−/− mice.10 However, excessive BA accumulation in the regenerating parenchyma can cause hepatocyte damage and disrupt normal hepatic regeneration.2 ,4 ,15 Therefore BA availability needs to be tightly regulated and the expression of cholesterol 7α-hydrolase (Cyp7a1), the rate-limiting enzyme in the classic pathway of BA synthesis, is a major control point.16 ,17 It is well known that after being drained into the intestine, BAs are actively absorbed and return to the liver through the enterohepatic circulation.16 ,18 Importantly, when the enterohepatic circulation of BAs is interrupted, for instance by external biliary drainage or by feeding a BA sequestering resin, liver regeneration after PH is impaired.10 ,19 However, short-term feeding of a non-toxic cholic acid (CA)-enriched diet is sufficient to elicit liver growth in the absence of tissue resection.10 ,13 These findings suggest that BA could be among the portal blood constituents that were postulated almost 50 years ago as essential factors in promoting liver regeneration.8 Interestingly, enterohepatic circulation of BA also implies the release to the portal blood of fibroblast growth factor 19 (FGF19 or Fgf15 in rodents).20 ,21 Fgf15 is an enterokine synthesised in the distal ileum after postprandial uptake of BAs by the enterocytes. After reaching the liver through the portal circulation, Fgf15 was shown to bind and activate the FGF receptor 4 (FGFR4), leading to the downregulation of Cyp7a1 expression and therefore to the inhibition of BA synthesis by hepatocytes.20 ,21 More recently, Fgf15 has been reported to have important metabolic effects on liver protein and glycogen metabolism, similar in some aspects to those of insulin.22 ,23
Together, these observations suggest that Fgf15 could be part of an enterohepatic endocrine circuit participating in liver regeneration. In this work we have directly addressed this issue. First we provide evidence showing that Fgf15 is essential for the preservation of BA homeostasis and consequently for liver regeneration to proceed normally. We also show that Fgf15 administration improves the outcome of cholestatic and small remnant livers after PH, suggesting its potential therapeutic value.1 ,2 ,24–26 Furthermore, we demonstrate that Fgf15 expression plays a key role in the well established effects of BA on liver growth.10 ,13
Methods
Animals
Fgf15 −/− mice and the wild type littermate controls (Fgf15 +/+) are on a mixed C57BL/6/129/Sv background, and have been backcrossed for more than 14 generations. These mice have been described before27 and were generously provided by Dr Rubinstein (UCSF, San Francisco, California, USA). Experiments were performed in male mice 8–12 weeks of age. Mice were housed under controlled conditions with 12 h light/dark cycles. Animals received humane care, and all experiments were carried out in compliance with our institution's ethical guidelines.
PH model
Liver regeneration was induced by 70% PH as described.13 ,28 For 85% PH all lobes were excised except the superior right lobe. Sham operation of mice was also performed. One hour prior to sacrifice, mice were intraperitoneally injected with 5′-Br-2′-deoxyuridine (BrdU) from Sigma (St Louis, Missouri, USA). When indicated, 6 days prior to PH, mice were fed with the standard diet supplemented with 2% cholestyramine resin (Sigma), and 5 days prior to PH, mice were infected with an Fgf15-expressing adenovirus or a control adenovirus (2×109 particles/g body weight, by retroorbital injection). These adenoviruses were a generous gift of Dr Mangelsdorf (University of Texas, Dallas, Texas, USA), and have been previously described.21 At the indicated time points portal and/or peripheral blood was obtained and mice were sacrificed by cervical dislocation. Liver tissue samples were snap frozen or formalin fixed and paraffin embedded.
Additional Materials and Methods are included as online supplementary files.
Results
Liver regeneration in Fgf15−/− mice after PH
To directly evaluate whether Fgf15 could have a role in liver regeneration we examined the response of Fgf15 −/− mice to 70% PH. The first phenotypic trait of these mice that captured our attention was their elevated mortality in the first 48 h after liver resection (figure 1A). The markedly reduced survival was accompanied by elevated levels of circulating liver enzymes and bilirubin (figure 1B), suggestive of enhanced liver injury in surviving Fgf15 −/− mice after hepatectomy. This was corroborated upon comparative histological examination of the remaining liver tissues of Fgf15 −/− and Fgf15 +/+ mice at different time points post PH (figure 1C). Quantitative analysis of tissue necrosis revealed significantly increased tissue damage in mice lacking Fgf15 (figure 1C). Moreover, staining with antibodies against cytokeratins 8 and 19 showed an increased cellular injury of both hepatocytes and cholangiocytes in Fgf15 −/− mice (see online supplementary figure S1).

Fgf15 −/− mice show increased liver injury and mortality after partial hepatectomy (PH). Survival of Fgf15 +/+ and Fgf15 −/− mice after 70% PH (n=10 mice per group) (A). Serum levels of alkaline phosphatise (ALP), alanine transaminase (ALT), aspartate transaminase (AST) and bilirrubin in Fgf15 +/+ and Fgf15 −/− mice at different time points after PH. *p<0.05 and **p<0.01 versus Fgf15 +/+ mice at matched time points (B). Representative images of liver tissue sections (H&E staining) from Fgf15 +/+ and Fgf15 −/− mice at different time points after PH. Graph shows the quantitative analysis of necrotic lesions (C).
We analysed the liver growth response and related parameters following PH. As shown in figure 2A, we found significantly slower regrowth of the liver in Fgf15 −/− mice at early time points after PH. At later stages (5–7 days), those Fgf15 −/− animals that survived the intervention showed similar liver weights to wild type mice (not shown). In agreement with this delayed regenerative response, we also observed an impaired expression of the cell cycle regulatory genes Cyclin-D1, proliferating cell nuclear antigen, Foxm1b and Cdc25b (figure 2B,C).5 ,7 ,10 ,13 ,29 The recovery in the expression of Cdc25b, a transcriptional target of Foxm1b,30 observed in Fgf15 −/− mice at 48 h post PH could be due to activation of compensatory mechanisms known to induce Cdc25b gene expression. These may include the hedgehog-inducible glioblastoma Gli-1 transcription factor and the epidermal growth factor receptor (EGFR) signalling system,31–33 as suggested by the increased levels of the mRNAs for Gli-1 and two EGFR ligands (amphiregulin and heparin-binding EGF) found in these animals (see online supplementary figure S2). Additionally, the expression of hepatocyte growth factor (HGF), a key mediator of liver regeneration,5–7 ,9 was reduced at early time points after PH in Fgf15 −/− mice, showing a delayed upregulation in these animals (see online supplementary figure S2). Interestingly, we also observed a significant downregulation of FXR mRNA levels in Fgf15 −/− mice after PH (see online supplementary figure S2). This could explain in part the profound reduction in the expression of its transcriptional target Foxm1b 34 observed in these animals following liver resection. Next, we evaluated the DNA synthetic response by immunohistochemical detection of BrdU incorporation. Consistent with the decreased expression of cell cycle-related genes, we found that the proportion of hepatocytes and bile duct epithelial cells in S phase was significantly reduced in Fgf15 −/− mice compared with wild types at various time points after PH (figure 2D). In agreement with these findings, the appearance of mitotic figures in hepatocytes after PH was also delayed in Fgf15 −/− mice (see online supplementary figure S3). Altogether, the impaired regenerative response of mice lacking Fgf15 was similar to that presented by FXR−/− mice, albeit mortality of Fgf15 −/− animals was significantly higher (∼70% vs ∼30%).10
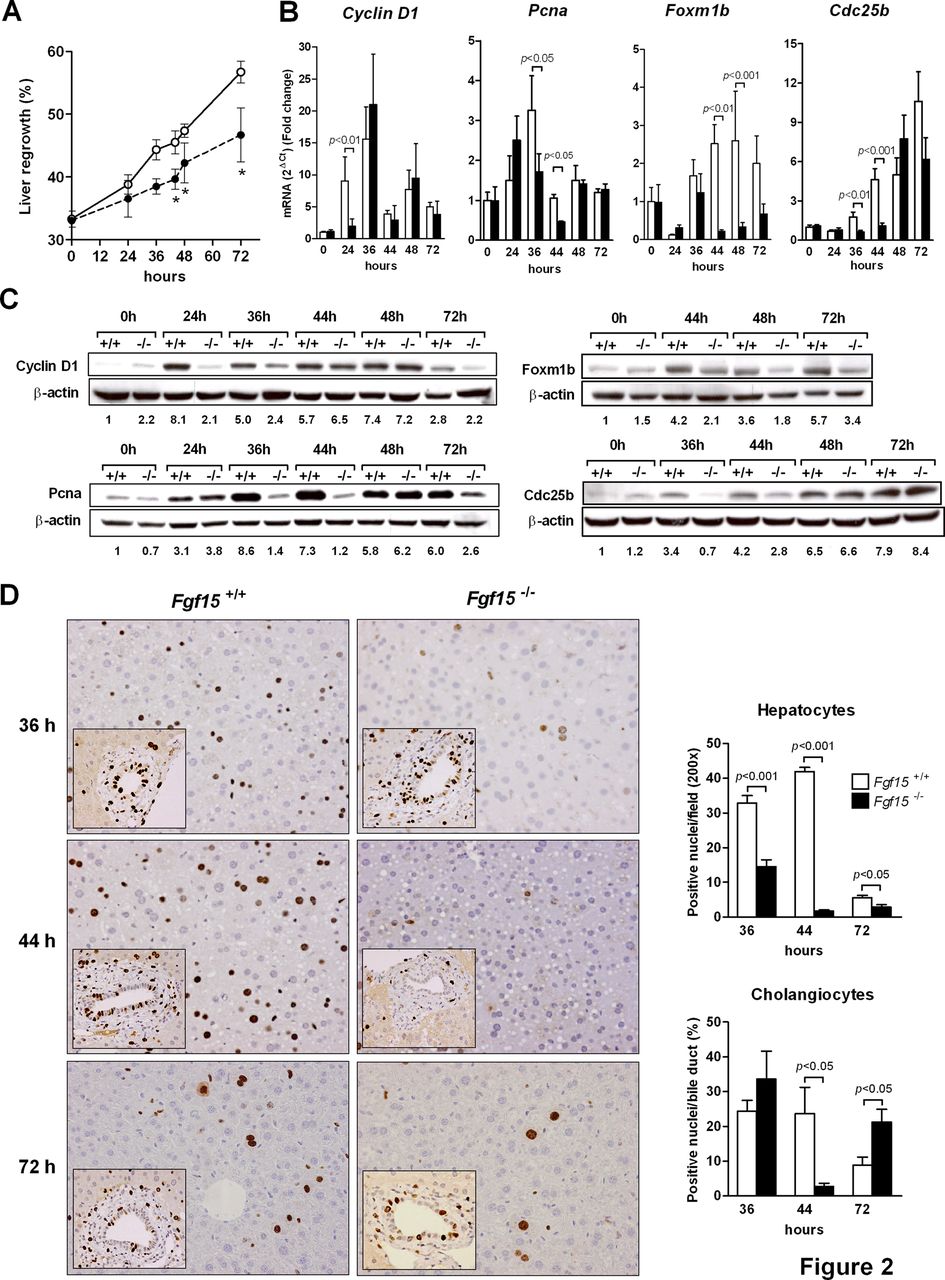
Liver regeneration after partial hepatectomy is defective in Fgf15 −/− mice. The extent of liver regeneration was assessed at different time points by measuring liver regrowth, *p<0.05 versus Fgf15 +/+ (A). The expression of proliferation-related genes at the mRNA (B) and protein level (C) was also evaluated; representative blots are shown and quantitation of band intensities (normalised to β actin) is indicated. DNA synthesis in hepatocytes and bile duct epithelial cells was assessed by immunohistochemical evaluation of BrdU incorporation into DNA (D).
BA homeostasis regulated by Fgf15 is essential for liver protection during regeneration
The mechanism accounting for the reduced survival of FXR−/− mice after PH has not been specifically addressed in previous studies.10 In view of the overt phenotype we observed in the Fgf15-deficient mice we decided to explore the potential underlying mechanisms. One of the key metabolic effects of Fgf15 is its ability to downregulate Cyp7a1 expression in hepatocytes, and therefore to reduce de novo synthesis of BA by the liver.21 ,23 Fgf15 −/− mice were shown to display increased basal expression of Cyp7a1 and elevated faecal BA excretion, suggestive of increased BA pool size.21 We corroborated here the marked upregulation of Cyp7a1 in the liver of Fgf15-deficient mice (figure 3A). Consistently, in Fgf15−/− mice, the BA pool size was significantly higher (figure 3B). Moreover, under basal conditions, intrahepatic BA levels were also noticeably elevated (figure 3C). To prevent liver injury and allow a normal regenerative response after PH, the control of intrahepatic BA levels, and in particular the suppression of BA synthesis through Cyp7a1 downregulation, is very important.15 ,25 Therefore we evaluated this issue by determining the time course of intrahepatic BA levels after PH. Following PH, Fgf15 −/− mice displayed a fast increase in intrahepatic BA concentrations, which remained higher until the last time point studied, when intrahepatic BA concentrations in wild type mice have already decreased to values close to preoperative levels (figure 4A). In agreement with previous reports,10 ,15 liver Cyp7a1 mRNA levels were clearly reduced shortly after PH in both genotypes, though they were still clearly detectable in Fgf15 −/− animals 6 and 12 h after liver resection (figure 4B). Accordingly, Cyp7a1 protein levels were also significantly higher in Fgf15 −/− mice (figure 4C). Interestingly, the expression of the chemokine macrophage inflammatory protein-2 (Mip-2), which is upregulated by BAs and has been recently involved in cholestasis-induced liver injury,35 ,36 was much higher in the livers of Fgf15 −/− mice after PH (see online supplementary figure S4).
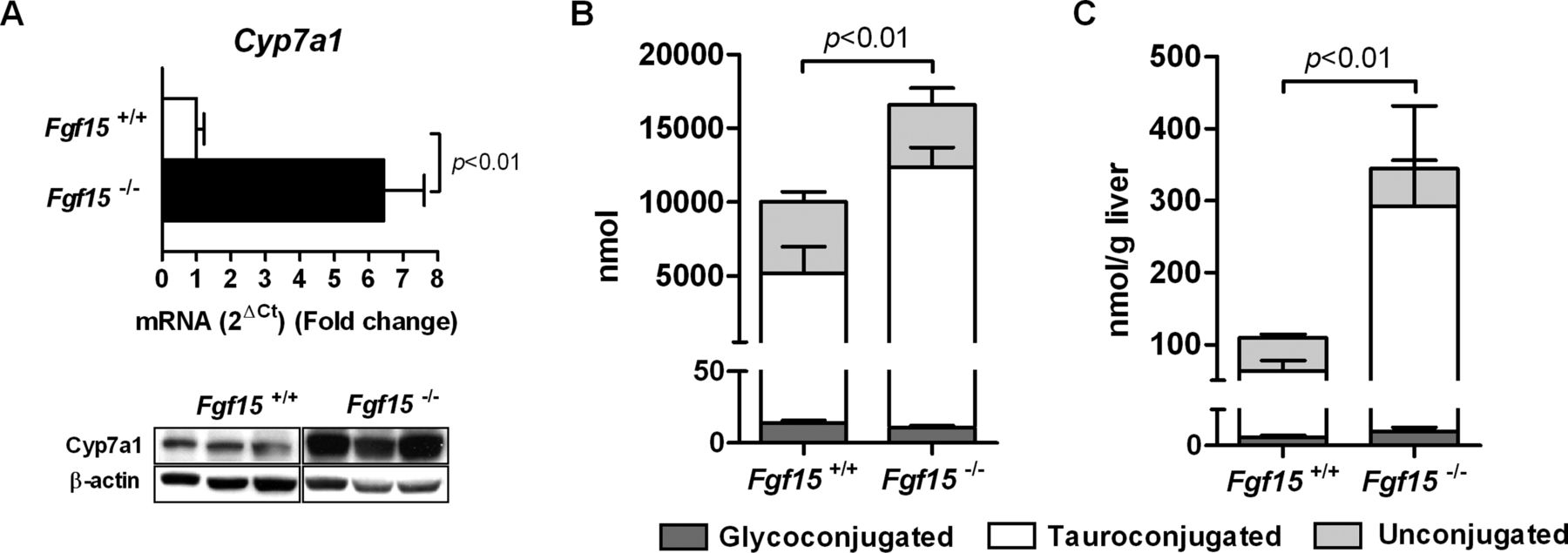
Fgf15 −/− mice display increased hepatic cholesterol 7α-hydrolase (Cyp7a1) expression and bile acid (BA) levels. Expression of Cyp7a1 mRNA and protein in the liver of wild type and Fgf15 −/− mice under basal conditions as measured by quantitative PCR and Western blotting respectively (A). Determination of the total BA pool size (B) and intrahepatic BA levels (C) in Fgf15 +/+ and Fgf15 −/− mice (n=5 mice per group).

Bile acid (BA) homeostasis is dysregulated in Fgf15 −/− mice after partial hepatectomy (PH). Intrahepatic BA concentrations were measured in wild type and Fgf15 −/− mice at the indicated time points after PH (A) (n=5 mice per group), *p<0.05. Expression of cholesterol 7α-hydrolase (Cyp7a1) mRNA (B) and protein (C) in wild type and Fgf15 −/− mice after PH as determined by quantitative PCR and Western blotting respectively.
Altogether, these observations suggested that the increased hepatic injury and consequent elevated mortality observed in Fgf15 −/− mice after liver resection could be due to BA-induced toxicity. To directly address this possibility we fed Fgf15 +/+ and Fgf15 −/− mice a diet supplemented with the BA-sequestering resin cholestyramine for 6 days, which leads to increased faecal output of BA.10 As shown in figure 5A,B, this diet significantly reduced portal and intrahepatic BA levels in Fgf15 −/− mice, bringing those levels down, close to the normal levels measured in wild types. Next we carried out PH in cholestyramine-fed mice, and found that lowering BA levels had a significant protective effect on Fgf15 −/− mouse survival (figure 5C). Upon resin feeding the surviving Fgf15 −/− mice showed a regeneration profile virtually indistinguishable from that of wild type animals also fed with cholestyramine, which in agreement with previous publications,10 was attenuated compared with animals fed the normal diet (figure 5D).

Downregulation of the bile acid (BA) pool by feeding a BA-sequestering resin protects Fgf15 −/− mice after partial hepatectomy (PH). Wild type and Fgf15 −/− mice were fed the standard diet or this same diet supplemented with 2% cholestyramine (2% resin) for 6 days, and the total BA concentrations were measured in portal blood serum (A) or in liver tissues (B). Survival after PH was monitored in Fgf15 +/+ and Fgf15 −/− mice fed the control diet or the cholestyramine-supplemented diet (n=6 mice per group) (C). Liver regrowth after PH was assessed in cholestyramine-fed Fgf15 +/+ (open triangles) and Fgf15 −/− (closed triangles) mice (n=6). Liver regrowth in Fgf15 +/+ mice fed control diet (open circles) is shown for comparison. *p<0.05 versus Fgf15 +/+ mice fed the cholestyramine diet (D).
These findings supported the notion that excessive BA levels could be determinant for the impaired regeneration and lethal outcome presented in these mice. In cases of biliary obstruction, biliary drainage has been reported to improve liver function tests.4 But lack of enterohepatic circulation, or external biliary drainage, were previously shown to prevent liver regeneration.25 With this in mind, and to further evaluate the protective role of Fgf15 in liver regeneration in the face of high BA concentrations, Fgf15 −/− mice were infected with a recombinant adenovirus that expresses Fgf15 or a control adenovirus. Five days after adenoviral infections we detected intrahepatic expression of Fgf15 (figure 6A), and confirmed its biological activity by showing reduced expression of Cyp7a1 mRNA (figure 6B). This effect translated into a significant reduction of intrahepatic BA levels, although these did not reach the values found in wild type mice (figure 6C). Thus we performed PH in Fgf15 −/− mice infected with control or Fgf15-expressing adenoviruses. As shown in figure 6D, expression of Fgf15 in the latter significantly improved survival.

Fgf15 delivery to the liver in Fgf15 −/− mice by an adenoviral vector reduces intrahepatic bile acid (BA) levels and increases survival after partial hepatectomy (PH). Fgf15 −/− mice were infected with a control adenovirus (Ad-Control) or an Fgf15-expressing adenovirus (Ad-Fgf15). Five days later Fgf15 (A) and cholesterol 7α-hydrolase (Cyp7a1) (B) gene expression levels were measured in the liver of these mice by quantitative PCR. Intrahepatic BA levels were also measured in Ad-Control and Ad-Fgf15-infected mice. Basal BA levels in the livers of uninfected Fgf15 +/+ and Fgf15 −/− mice are also shown for comparison (C). Survival after PH of Fgf15 −/− mice infected with Ad-Control or Ad-Fgf15 5 days prior to liver resection (n=6 mice per group) (D).
Fgf15 delivery increases survival of wild type mice after extensive liver resection
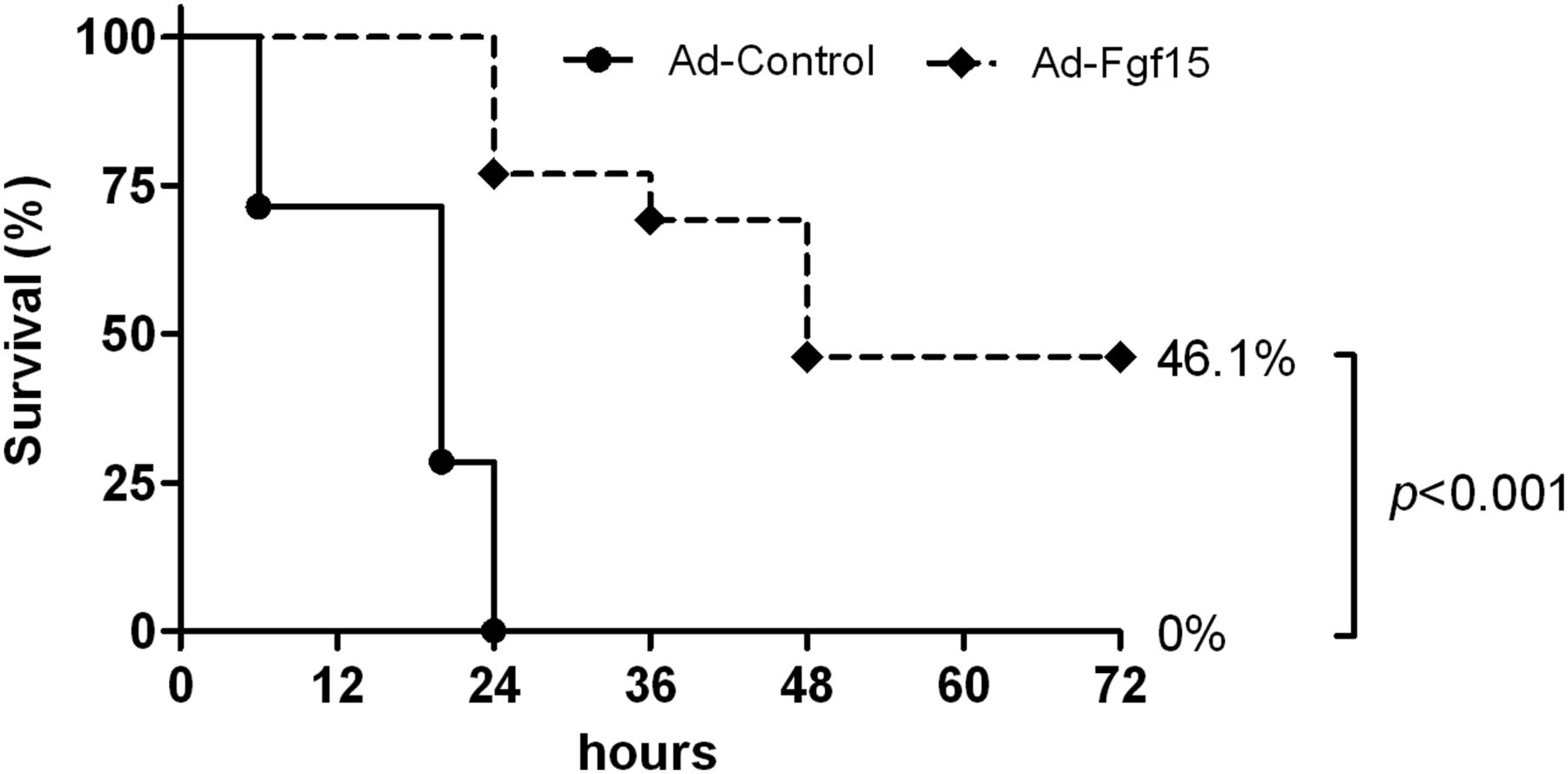
We also evaluated the protective effects of Fgf15 in a model of acute liver failure due to extensive parenchymal resection (85% PH) in wild type mice. In this model of the small-for-size syndrome (SFSS) we found that all animals infected with the control adenovirus died within the first 24 h post hepatectomy. However, mice that were infected with the Fgf15-expressing virus showed significantly increased survival (figure 7). In line with this finding we observed that the intrahepatic expression of the pro-regenerative growth factor HGF was significantly increased in mice infected with the Fgf15-expressing virus, while the expression levels of the inflammatory chemokine Mip-2 were downregulated (see online supplementary figure S5A,B).

Fgf15 delivery to the liver improves the survival of wild type mice after extensive liver resection. Wild type mice were infected with a control adenovirus (Ad-Control) or an Fgf15-expressing adenovirus (Ad-Fgf15). Five days later all mice underwent extensive hepatectomy (85%) and survival was monitored. (n=13 mice per group).
Fgf15 is required for BA-induced liver growth
The findings described above showed the critical role of Fgf15 in the preservation of BA homeostasis to prevent liver injury after PH. Additionally, a potential participation of Fgf15 in the mechanisms of the regenerative processes controlled by BA may be considered. To address this possibility we fed Fgf15 +/+ and Fgf15 −/− animals a diet supplemented with CA (1%) for 5 days, which has been shown to promote liver growth in mice in the absence of parenchymal resection.10 ,13 We found that Fgf15 gene expression was induced in the ileum of wild type mice (figure 8A), which was accompanied by the expected increase in liver mass (figure 8B). However, the growth response was blunted in Fgf15 −/− animals (figure 8B). It is noteworthy that after CA feeding Fgf15 −/− mice showed elevated levels of circulating liver enzymes compared with wild type mice (figure 8C), indicative of increased liver injury. In agreement with the impaired liver growth response of Fgf15 −/− mice, we found that staining with the nuclear antigen Ki-67, present in proliferating cells,37 was significantly reduced in the hepatocytes of these mice (figure 8D). Interestingly, Ki-67 positivity was also markedly low in bile duct epithelial cells (cholangiocytes) from Fgf15 −/− animals fed the CA-supplemented diet (figure 8D). Consistent with these data was the impaired hepatic expression of the transcription factor Foxm1b and its direct target Cdc25 (figure 8E), which are known to be induced during liver growth upon CA-diet feeding.10
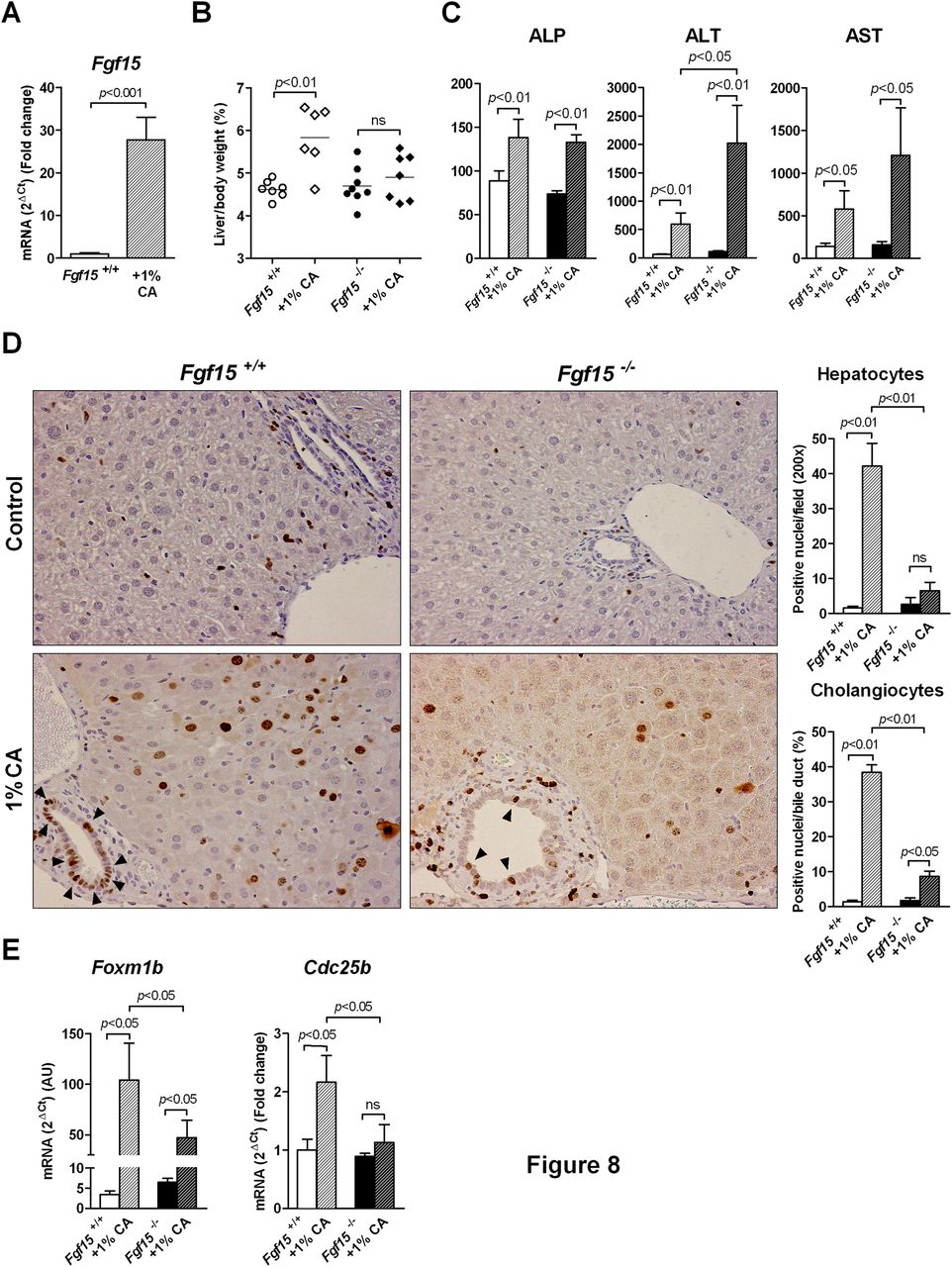
Bile acid induced liver growth and proliferation of liver epithelial cells is impaired in Fgf15 −/− mice. Feeding a diet supplemented with 1% cholic acid (CA) for 5 days induces Fgf15 gene expression in the ileum of wild type mice (A), and differentially stimulates liver growth in Fgf15 +/+ and Fgf15 −/− mice (B). Serum levels of alkaline phosphatase (ALP), alanine transaminase (ALT) and aspartate transaminase (AST) in wild type and Fgf15 −/− mice fed control or CA supplemented diets for 5 days (C). Cellular proliferation stimulated by CA feeding as determined by immunohistochemical detection of the proliferation marker Ki-67. Representative stainings are shown, and quantitation of positive hepatocytes and biliary epithelial cells (cholangiocytes) (arrow heads) are depicted in graphs (D) (n=7 mice per group). Expression of proliferation-related genes in the liver of Fgf15 +/+ and Fgf15 −/− mice fed control or CA supplemented diets as determined by quantitative PCR (E).
In view of these findings we next examined whether Fgf15 could behave as a direct mitogen for mouse hepatocytes and cholangiocytes. As depicted in figure 9A these cells express FGFR4 and β-Klotho, the receptor and coreceptor of Fgf15 respectively.23 Stimulation with FGF19 induced the activation of the mitogen-activated protein kinase signalling pathway, as indicated by increased phosphorylation of Erk1/2 (figure 9B) and of the Erk1/2-kinase mitogen-activated protein kinase/extracellular signal-regulated kinase (see online supplementary figure S6A). FGF19 also induced DNA synthesis in primary hepatocytes and the proliferation of cultured mouse cholangiocytes to levels comparable to those triggered by the well established mitogens HGF and EGF (figure 9C). These findings were reproduced in the human hepatocyte cell line HepaRG (see online supplementary figure S6B) and in the murine cholangiocytic cell line BNL 1ME A.7R.1 (figure 9B,C). Interestingly, in cultured normal mouse cholangiocytes and BNL 1ME A.7R.1 cells we also observed an additive effect of FGF19 and CA on cell proliferation (see online supplementary figure 7A,B). Moreover, FGF19 stimulation also promoted a wound healing reaction in an in vitro scratch assay using AML12 mouse hepatocytes (see online supplementary figure S8), further supporting a role for this growth factor in liver regeneration and repair.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Treatment with fibroblast growth factor (FGF19) triggers intracellular signalling and proliferation of cultured mouse hepatocytes and biliary epithelial cells. Expression of Fgf15/FGF19 receptor FGFR4 and coreceptor β-Klotho was assessed by quantitative PCR in primary mouse hepatocytes and cultured cholangiocytes. Expression levels were normalised to those found in hepatocytes, which were given the arbitrary value of one (A). Mouse hepatocyes, cholangiocytes and BNL 1ME A.7R.1 cells were stimulated with recombinant FGF19 (rFGF19) for 10 min as indicated, and phosphorylated extracellular signal-regulated kinase1/2 (Erk1/2) and total Erk levels were determined by Western blotting. Representative blots of two experiments performed in duplicates are shown (B). Treatment with FGF19 stimulates DNA synthesis ((3H)-thymidine incorporation into DNA) in primary mouse hepatocytes, and the proliferation of cultured normal cholangiocytes and BNL 1ME A.7R.1 cells. Values were normalised to those found in control cultures (100%). The effects of hepatocyte growth factor (20 ng/ml) on hepatocyte DNA synthesis and epidermal growth factor (25 ng/ml) on cholangiocyte and BNL 1ME A.7R.1 cell proliferation are shown. *p<0.05, **p<0.01, ***p<0.001 (C).
Discussion
The influence of BA in the process of liver regeneration is complex and multifaceted. On the one hand elevated BA levels can be toxic for the liver parenchyma, and significant adaptations in the synthesis and transport of BA take place during liver regeneration to maintain adequate levels of these metabolites in the enterohepatic circulation.11 Clinical observations clearly indicate that patients with jaundice of different aetiology have a worse outcome after PH.2 ,4 This has been confirmed in surgical models of cholestasis and upon genetic manipulation of key proteins involved in BA homeostasis, such as Cyp7a1 and FXR.2 ,10 ,15 ,25 On the other hand, BAs are increasingly recognised as essential signalling molecules and mediators of liver regeneration.10 ,38 Insufficient BA synthesis, or their withdrawal from the enterohepatic circulation, quells liver growth after PH.10 ,19 ,39 Together these observations attest to the importance of fine-tuning BA levels to protect hepatocytes from BA-induced toxicity while allowing sufficient BA signalling for regeneration. In this work we have identified Fgf15 as an essential hormone in the modulation of BA levels during liver regeneration. In agreement with the key role played by Fgf15 in the inhibition of BA synthesis we found that under basal conditions Fgf15 −/− mice had an increased BA pool size. We also observed that mice lacking Fgf15 showed markedly elevated intrahepatic BA levels and a dramatic mortality rate after PH. Importantly, in the absence of Fgf15 the rapid downregulation of Cyp7a1 expression that occurs after PH, which is critical to lower liver BA levels in the regenerating liver,15 was less efficient. The contribution of toxic BA concentrations to the exacerbated mortality of Fgf15 −/− animals after liver resection was demonstrated by the protective effect of cholestyramine feeding, which effectively lowered portal blood and intrahepatic BA levels. Along these lines, an interesting recent report has demonstrated that transgenic expression of a constitutively active form of FXR in the intestine was able to reduce the BA pool size and to protect the liver from a variety of experimental cholestatic conditions.40 Our current findings agree with this study and complement it by supporting the notion that Fgf15 can be an important FXR target gene involved in the anticholestatic effects of activated FXR in the enterocyte.
In our study we have also shown the beneficial effects of infecting Fgf15 −/− mice with an Fgf15-expressing adenovirus prior to PH. Delivery of Fgf15 to the liver reduced intrahepatic BA concentrations and significantly improved survival after liver resection. These observations further confirm the protective effects of Fgf15 in our experimental setting, but perhaps most importantly advocate for a potential therapeutic application of controlled FGF19 administration to patients with cholestasis undergoing major liver resection or liver transplantation.2 ,4 ,24 Preoperative biliary drainage has been applied in patients with obstructive jaundice, but this manoeuvre does not confer a survival benefit and may increase morbidity.2 ,4 ,41 Furthermore, experimental evidences indicate that in rats with obstructive jaundice preoperative external biliary drainage impairs liver regeneration after PH, a defect that is not observed upon internal biliary drainage into the stomach.19 While cholestasis may indeed impair liver regeneration, these findings substantiate the role of enterohepatic signals triggered by BA for the regenerative process, among which we believe Fgf15 is crucial. Interestingly, recent studies revealed that the expression of FGF19 (the human orthologue of Fgf15) is induced in the liver of patients with cholestasis and that BA can directly promote FGF19 upregulation in cultured human hepatocytes, which would be part of the feedback signalling loop to self-control BA pool size by reducing BA synthesis.42 ,43 This response may be regarded as an endogenous protective reaction of the liver towards cholestasis. All in all, our findings suggest that enhancement of this mechanism by the preoperative administration of recombinant FGF19 to patients with cholestasis could be a more physiological way to downregulate BA levels. Moreover, the increased survival after an extensive liver resection found in wild type mice infected with an Fgf15-expressing adenovirus points to the potential application of this factor to prevent liver failure after major hepatectomy. Enhanced survival of these mice was accompanied by increased intrahepatic expression of HGF, a key pro-regenerative factor,5–7 ,9 and reduced levels of the inflammatory chemokine Mip-2, which has been involved in different modes of acute liver injury.35 ,36 ,44 ,45 These responses are consistent with the recently reported growth-promoting and anti-inflammatory effects of FGF19,46 ,47 and suggest that this growth factor could be of interest in the management of the SFSS.1 ,4 ,39 ,48
Our study also provides additional insights suggesting the implication of Fgf15 in the basic mechanisms of liver regeneration. In addition to the exacerbated liver injury and the overt lethal phenotype of Fgf15 −/− mice after PH due to the build up of toxic BA levels, these animals exhibited signs of impaired regeneration. These included defective expression of proliferation-related genes and a delayed recovery of liver mass in surviving animals. Previous studies have shown that injection of recombinant FGF19 to mice resulted in increased hepatocyte proliferation.46 ,49 However, direct evidence for a mitogenic effect of FGF19/Fgf15 on liver cells has not yet been provided either in vitro or in vivo. To test this, we examined the growth-promoting effects of a CA-supplemented diet in wild type and Fgf15 −/− mice. This experimental approach avoided the severity of PH in Fgf15 −/− mice and also allowed us to directly test the contribution of gut-derived Fgf15 in BA-induced hepatocellular proliferation, since at variance with the situation found in humans, Fgf15 expression is not triggered by BAs in mouse hepatocytes.21 We found that CA feeding induced significantly less liver growth in Fgf15 −/− mice than in wild type mice. Although this CA diet showed little toxicity, Fgf15 −/− mice displayed enhanced liver injury compared with wild type animals. In spite of that, the compensatory growth response generally associated with liver injury29 ,50 was attenuated in these mice, suggesting the specific impairment of important endogenous mechanisms. The differential liver growth between the two mouse genotypes was likely due to a decreased progression through the cell cycle in the targeted mice, as indicated by Ki-67 staining. Interestingly, and consistent with our observations in the PH model, reduced Ki-67 staining was found in hepatocytes an in bile duct epithelial cells of Fgf15 −/− mice. Cholangiocyte proliferation upon BA feeding has been demonstrated before in rats.51 Our present findings indicate that in vivo Fgf15 would be an important mediator of BA-induced proliferation in liver parenchymal and biliary epithelial cells. In primary cultures both cell types were found to express FGFR4 and the FGF19 coreceptor β-Klotho,23 and FGF19 stimulation triggered intracellular signalling (Mek/Erk1/2 phosphorylation). Furthermore, our in vitro experiments clearly demonstrate that FGF19 is a direct mitogen for these two cell types, and that it may potentiate the mitogenic effects that BA exert on cholangiocytes.52
Collectively our results indicate that Fgf15 could behave as a gut-derived hormone that reaches the liver through the portal circulation fostering hepatic regeneration.5 ,8 Interestingly, besides its unique ability to modulate BA synthesis, recent studies have demonstrated that Fgf15 shares with insulin important effects on protein and glucose metabolism, even though both hormones exert these effects via distinct intracellular signalling pathways, suggesting a complementary mechanism of action.22 ,23 Certainly, these metabolic effects of Fgf15 may also be involved in its pro-regenerative activity. However, as we have demonstrated here, Fgf15 also exhibits a direct mitogenic action on liver cells which is not displayed by insulin.5 ,8 Therefore, it would be very interesting to examine the therapeutic value of FGF19 administration to patients undergoing liver resection, either alone as recently tested with insulin53 or in combination with the pancreatic hormone. Moreover, our findings also suggest that synthetic compounds that strongly activate FXR, and in turn can upregulate the expression of FGF19,34 ,54 could be useful to potentiate liver regeneration in patients with cholestasis undergoing major liver surgery. Indeed, FXR has been recently identified as a hepatoprotective receptor, and its pharmacological activation may promote liver repair and regeneration and compensate for its marked downregulation observed in models of liver injury55 and in our current study in Fgf15 −/− mice.
Acknowledgments
We thank Ms M Molina and M Mendez for their help with mouse hepatocyte isolations. Also we thank Drs R Hernandez-Alcoceba and M Gonzalez-Aparicio for their help with the preparation of adenoviral vectors.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 10 - Online figure 8
- Data supplement 11 - Online table 1
- Data supplement 2 - Online supplement
- Data supplement 3 - Online figure 1
- Data supplement 4 - Online figure 2
- Data supplement 5 - Online figure 3
- Data supplement 6 - Online figure 4
- Data supplement 7 - Online figure 5
- Data supplement 8 - Online figure 6
- Data supplement 9 - Online figure 7
Footnotes
-
Contributors IU, MGF-B, MUL, EV, HCYC, SC, UV-G, SM, MJM and ARC performed the experiments. IU, MGF-B, JFM, JJGM, MJM, CB, JP and MAA discussed and interpreted the data. MAA designed the experiments and wrote the manuscript. IU and MGF-B are co-first authors. CB, JP and MAA share senior authorship.
-
Funding This work was supported by the agreement between FIMA and the ‘UTE project CIMA’; RTICC-RD06 00200061 (CB, MAA), CIBEREhd (IU, MGFB, MJM, JJGM, JFM, JP), FIS PI10/02642 and PI10/00038 (CB, MAA), Ramón y Cajal Program (MUL), MICINN SAF2010-15517 (MJM, JJGM) and SAF2009-11538 (JFM), Junta de Castilla y León Biomedicina-2011/JDI6 and SA023A11-2 (MJM, JJGM), and Fundación Condesa de Fenosa (JP).
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.