Article Text

Abstract
Objective The differential role of the IL-1 agonists, IL-1α, which is mainly cell-associated versus IL-1β, which is mostly secreted, was studied in colon inflammation.
Design Dextran sodium sulfate (DSS) colitis was induced in mice globally deficient in either IL-1α or IL-1β, and in wild-type mice, or in mice with conditional deletion of IL-1α in intestinal epithelial cells (IECs). Bone marrow transplantation experiments were performed to assess the role of IL-1α or IL-1β of myeloid versus colon non-hematopoietic cells in inflammation and repair in acute colitis.
Results IL-1α released from damaged IECs acts as an alarmin by initiating and propagating colon inflammation, as IL-1α deficient mice exhibited mild disease symptoms with improved recovery. IL-1β is involved in repair of IECs and reconstitution of the epithelial barrier during the resolution of colitis; its deficiency correlates with disease exacerbation. Neutralisation of IL-1α in control mice during acute colitis led to alleviation of clinical and histological manifestations, whereas treatment with rIL-1Ra or anti-IL-1β antibodies was not effective. Repair after colitis correlated with accumulation of CD8 and regulatory T cells in damaged crypts.
Conclusions The role of IL-1α and IL-1β differs in DSS-induced colitis in that IL-1α, mainly of colon epithelial cells is inflammatory, whereas IL-1β, mainly of myeloid cell origin, promotes healing and repair. Given the dissimilar functions of each IL-1 agonistic molecule, an IL-1 receptor blockade would not be as therapeutically effective as specific neutralising of IL-1α, which leaves IL-1β function intact.
- INTESTINAL EPITHELIUM
- EXPERIMENTAL COLITIS
- GUT INFLAMMATION
- INTERLEUKINS
- MUCOSAL IMMUNITY
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
-
Expression of IL-1 in inflamed colon tissue has been reported in patients with inflammatory bowel disease (IBD) and in experimental colitis. However, no studies have assessed in parallel the role of IL-1α versus IL-1β in colon inflammation and repair.
What are the new findings?
-
The IL-1α precursor, which is mainly released from colon epithelial cells during acute colitis, is the major IL-1 molecule that mediates colon inflammation, acting initially as an alarmin. IL-1α from infiltrating bone marrow-derived cells amplifies and sustains the inflammatory response.
-
IL-1β plays a dominant role in colon repair in colitis, by affecting epithelial cell proliferation and restoration of the colon barrier.
-
T cell infiltration into damaged crypts, such as regulatory and CD8 T cells, correlates with colon repair.
-
Treatment with anti-IL-1α antibodies is effective in amelioration of dextran sodium sulfate-induced colitis.
How might it impact on clinical practice in the foreseeable future?
-
Understanding the differential role of IL-1α and IL-1β in colitis and its progression to colorectal cancer may lead to treatment of patients with IBD with neutralising antibodies to IL-1α. Blocking the IL-1 receptor may reduce the healing benefits of IL-1β.
Introduction
Inflammatory bowel disease (IBD) is characterised by recurrent inflammation of the gastrointestinal tract with increased risk of developing colorectal cancer (reviewed in refs. 1–5 1–5). The pathological features of IBD include injury of intestinal epithelial cells (IEC) with disruption of the mucosa, which provides a barrier between the intestinal microflora and the host's internal organs. With loss of barrier function, there is subsequent inflammation induced by bacterial invasion into the lamina propria. The integrity of the mucosal epithelium is dependent on interactions between the microbiota and pathogen recognition receptors expressed in IECs, as well as cytokines and chemokines from intestinal cells. IECs also contribute to innate immunity in the gut by secreting antimicrobial peptides such as defensins (reviewed in refs. 6–11 6–11).
Cells of the innate and adaptive immune systems and a diversity of cytokines/chemokines contribute to IBD; however, the specific role each plays in the pathogenesis of disease remains unclear (reviewed in refs. 7 9–157 ,9–15). Of special relevance to colon inflammation is IL-1, which initiates and propagates inflammation (reviewed in refs. 16–18 16–18). The two major IL-1 agonists, IL-1α and IL-1β, are synthesised as precursors. IL-1β is not constitutively present in healthy subjects. Upon inflammatory stimuli, IL-1β is secreted in its mature form after processing in the inflammasome by caspase-1. The IL-1α precursor is constitutively present in homeostatic conditions in epithelial and mesenchymal cells, is biologically active, and signals through binding to the IL-1 receptor. The IL-1α precursor is processed by calpain to generate the mature active IL-1α form. A membrane-associated form of IL-1α, through which IL-1R-positive cells can be activated in a juxtacrine manner, also exists. We previously showed that in vivo, IL-1β and IL-1α differ in their compartmentalisation within the producing cell or its microenvironment, resulting in distinct functions (reviewed in refs. 16 1716 ,17). The intracellular IL-1α precursor can translocate to the nucleus, especially upon stress or inflammatory stimuli, and controls gene expression, cell proliferation and differentiation.16 ,17 ,19 ,20 Cell-associated IL-1α functions as an alarmin when the precursor is released along with cell contents from necrotic cells and initiates inflammation by recruiting neutrophils.21 Subsequently, IL-1β is released in its mature form from recruited macrophages, and propagates the inflammatory response.19 ,21
Extensive expression of IL-1 in colons of IBD patients and involvement of IL-1 molecules in the pathogenesis of colitis has been described.22–26 Nevertheless, which of the two IL-1s play a direct role in initiating and perpetuating colitis has not been evaluated. To delineate the role IL-1α and IL-1β in the development and repair of acute colon inflammation, we used the dextran sodium sulfate (DSS)-induced colitis model.
Methods
Mice
C57BL/6 wild type (WT) mice were purchased from Harlan Laboratories (Rehovot, Israel). IL-1α and IL-1β knockout (KO) mice were generated in the laboratory of Prof Y Iwakura. These mice were backcrossed to C57BL/6 mice for more than eight generations and are homozygous for the relevant mutation. Mice were bred and kept at the Animal Facilities of the Faculty of Health Sciences, Ben-Gurion University, Beer-Sheva, Israel. IL-1αloxP mice were generated by the Taconic Artemis Company, (Cologne, Germany) by introducing flanked loxP sites between the coding exons 2–5. To generate mice with an intestine-specific deletion of IL-1α, Villin-Cre mice, which were kindly obtained from Prof Y Ben-Nerriah, Hebrew University, Jerusalem, Israel, were mated with IL-1αloxP mice. Genotyping of mice with conditional depletion of IL-1α in epithelial cells (IL-1αint/Δ) was performed by PCR, using genomic DNA isolated from the tail by the REDEXTRACT-N-AMP tissue kit (Sigma, Israel). Cre recombinase activity was induced by injection of tamoxifen (200 mg/kg body weight, dissolved in corn oil, Sigma), on three consecutive days. There was no detectable background recombination in the presence of the Villin-Cre-ERT2 transgene, and no phenotypic differences between these strains. Animal studies were approved by the Animal Care Committee of Ben-Gurion University.
DSS-induced colitis model
A solution of 2.5% DSS (36–50 000 kDa, MP Biomedicals, Solon, Ohio, USA) was administered in the drinking water for 7 days and subsequently replaced with regular water. Clinical signs of colitis were recorded daily, and the disease activity index (DAI), which includes weight loss, stool consistency and occult blood in the stool, was determined. Occult/gross bleeding was measured using the Cenogenics Stool Blood Test (No SB-21, Cenogenics Corporation, Morganville, New Jersey, USA). Weight loss was calculated as the percentage change compared with the body weight before initiation of the experiment. Colon length from the rectum to the caecum was measured at the endpoint of experiments.
In vivo permeability assay
Permeability of fluorescein isothiocyanate dextran (FITC-D, MW 4 000; Sigma) was assessed, as described.27
Histological studies
Colons were excised on days 0, 8 or 15 after beginning DSS treatment and embedded in paraffin according to a standard protocol. The severity of colitis was assessed in a blind fashion by a pathologist in H&E stained sections as described.10 The number of lymphoid follicles (LF) was counted in H&E stained sections (the mean number of LF in six random fields).
Immunohistochemistry and immunofluorescence staining
The following primary antibodies were used: anti-proliferating cell nuclear antigen (PCNA), (Dako Cytomation, Carpinteria, California, USA); polyclonal rabbit anti-claudin-3 (Acris Antibodies, San Diego, California, USA); rabbit polyclonal antimyeloperoxidase (MPO) (Abcam, Cambridge, UK), mouse monoclonal CD4 (Abcam), polyclonal rabbit anti-mouse CD8 (Novus Biologicals, Littleton, Colorado, USA), rat anti-human CD3, (Serotec, Oxford, UK), mouse anti-mouse FOXP3 (BioLegend, San Diego, California, USA), goat anti-IL-1β (R&D Systems), goat anti-IL-1α (R&D Systems) and anti-cytokeratin-18 (Santa Cruz Biotechnology, Santa Cruz, California, USA).
The Vectastain Elite ABC Peroxidase kit or Universal ImmPRESS kit (Vector Laboratories, Burlingame, California, USA) was used as secondary antibodies, and visualisation was performed using 3-amino-9-ethylcarbazole (AEC) as a substrate (ZYMED Laboratories, San Francisco, California, USA). The number of CD3-, CD4-, CD8-, CD25-, FoxP3-, PCNA- or MPO-positive cells was counted in stained sections in six randomly chosen fields (×400), and results are presented as an average.
For immunofluorescence studies, secondary antibodies were conjugated with cy-2, cy-3 or cy-5 (Jackson Immuno Research, West Grove, Pennsylvania, USA). Sections were examined under a Zeiss Laser Scanning Confocal Microscope.
Generation of bone marrow (BM) chimeric mice
Recipient WT, IL-1α and IL-1β KO mice were irradiated with 13 Gy of Cobalt 60 total. One day later, BM cells from the femur and tibia of WT/GFPtransgenic mice were injected intravenous via the retro-orbital plexus (5×106 cells in 200 μL of PBS/mouse). Mice received Baytril antibiotics (Bayer, Leverkusen, Germany), 5% in drinking water for 45 days and after 60 days DSS treatment was administered.
In vivo IL-1 netralisation
WT mice were treated with DSS, as described, or with anti-IL-1α (eBioscience, San Diego, California, 10 µg/mouse). Antibodies were injected on days 0, 4 and 6. rIL-1Ra (50 mg/kg, Amgen, Thousand Oaks, California, USA) was injected 2 days before treatment, and every day after DSS administration, until day 6.
Statistical analysis
Each experiment was performed 2–6 times, and each experimental group consisted of 6–12 mice. Data are expressed as mean±SEM. Differences were analysed using the Student t test and the one-way ANOVA test. Survival curves were calculated using the Kaplan–Meier method.
Results
IL-1α deficiency correlates with a mild form of colitis and enhanced tissue repair, while IL-1β deficient mice exhibit a severe and non-healing disease
IL-1α KO mice were more resistant to DSS-induced colitis than WT or IL-1β KO mice and escaped the lethal consequences of DSS treatment observed in IL-1β deficient mice (figure 1A). In WT mice, and to a lesser extent in IL-1α deficient mice, the DAI increased during the 7 days of DSS administration but quickly decreased upon termination of DSS treatment. By contrast, in IL-1β KO mice, body weight loss and severe bloody diarrhoea from day 6 onward were more pronounced, and the DAI continued to increase, even after cessation of DSS treatment (figure 1B). In WT and IL-1β KO mice treated with DSS, but not in IL-1α KO mice, we observed shortening of the colon compared with untreated mice (figure 1C, D).

Differences between IL-1α and IL-1β deficient mice in response to dextran sodium sulfate (DSS) colitis. (A) Survival of IL-1α and IL-1β deficient mice compared to WT mice following 7 days of treatment with 2.5% DSS in the drinking water (10–12 mice/group). Differences in survival were calculated from six independent experiments. Survival curve was calculated using the Kaplan–Meier method. (B) Disease activity index was scored daily in IL-1α (9–13 mice/group), IL-1β (12–13 mice/group) and WT mice (11–15 mice/group) after 7 days of DSS treatment. Data represents mean±SEM from six independent experiments. p Values were calculated with ANOVA; *p<0.05, **p<0.001. (C) Lengths of colons after 7 days of DSS treatment. Pictures of one representative experiment out of six performed. (D) Mean±SEM colon length after 7 days of DSS treatment (5–8 mice/group). The data are from four independent experiments, p values were calculated with ANOVA; **p<0.001.
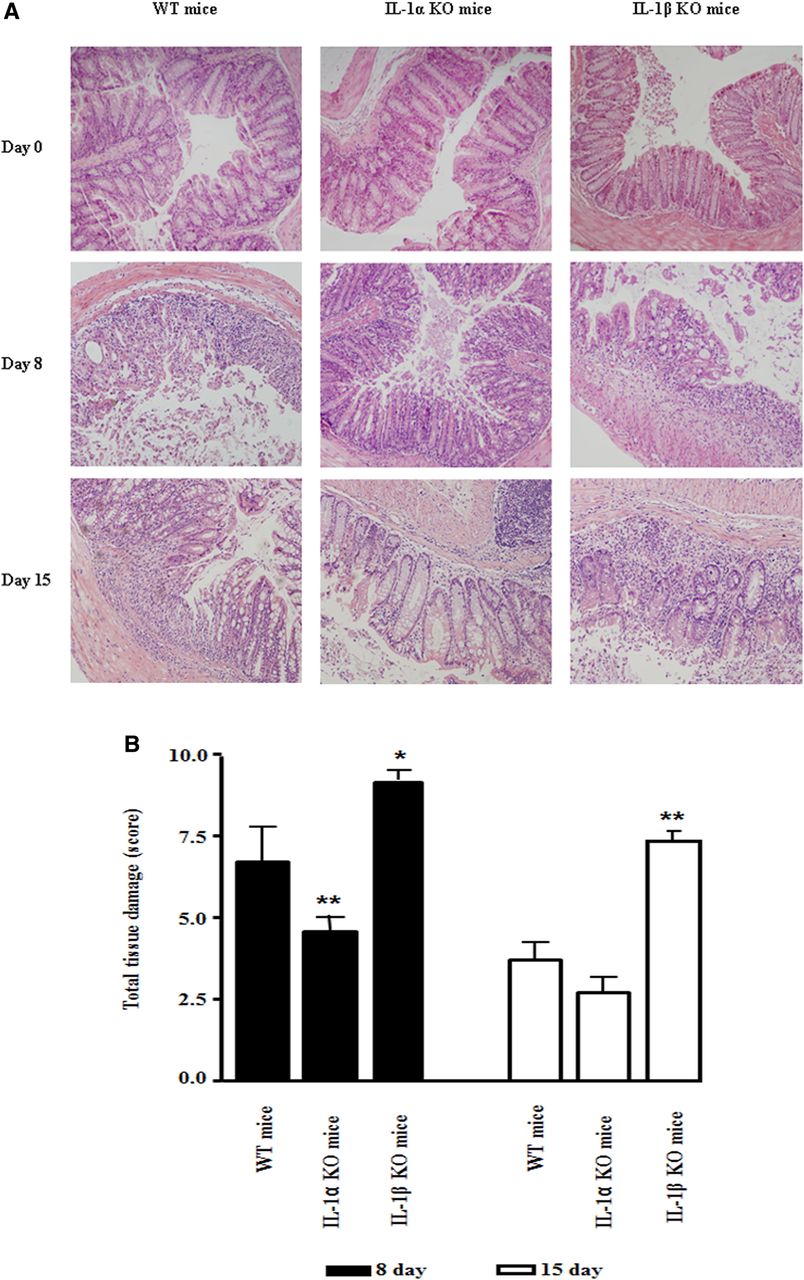
Histological studies of colon sections further validated the clinical observations (figure 2A). The structure of colon tissue before DSS treatment was similar in all groups. Thus, deficiency in IL-1α or IL-1β does not result in spontaneous colon inflammation. However, on day 8 of DSS treatment, tissue damage characterised by crypt loss, the presence of inflammatory cells, especially neutrophils and eosinophils, as well as areas of skip lesions, were observed in all DSS-treated mice. Nevertheless, tissue damage, including crypt loss, infiltration of inflammatory cells into the submucosa and muscularis was more severe in WT mice than in IL-1α deficient mice. In IL-1β KO mice, intensive crypt damage was found after the first days of DSS administration, with further exacerbation by day 8. In histological sections from mice on day 15, we noted only minimal inflammation and initial signs of colon tissue repair in WT mice, and almost complete repair in IL-1α KO mice; intensive inflammation with an influx of neutrophils and no signs of repair were observed in IL-1β KO mice. Semiquantitative scoring of the total histological index is shown (figure 2B).

Histological changes in colons of IL-1 deficient mice after dextran sodium sulfate (DSS) treatment. (A) Representative photomicrographs of colons of DSS-treated IL-1 deficient and WT mice, sacrificed on days 0, 8 and 15 (H&E staining, magnification ×200). (B) Mean±SEM histological score of colons on days 8 and 15 after the start of DSS treatment (8–12 mice/group). The data are from five independent experiments. p Values were calculated with ANOVA; *p<0.05 and **p<0.001.
Further studies were performed to evaluate the role of the IL-1 molecules in colon repair after cessation of DSS treatment. Staining of colon crypts with mouse PCNA showed that the proliferation rate of cells was higher in IL-1α KO mice compared with WT mice, and only minimal proliferation was seen in IL-1β deficient mice (figure 3A). To functionally assess the barrier function of the colon, we performed the in vivo permeability assay. In non-treated mice, only minimal levels of FITC-D were detected in the blood, whereas, in all DSS-treated mice, high levels of FITC-D were found. IL-1β KO mice showed maximal permeability that correlated with the most severe form of disease (figure 3B).
Colon repair was further demonstrated by immunostaining for the tight junction protein, claudin-3 in crypts (figure 3C). On day 8, in WT and IL-1β KO mice, where most crypts were destroyed, only a limited number of claudin-3-positive cells were found; whereas in IL-1α KO mice its expression was maintained. On day 15, preserved crypts were observed in WT mice, while the IEC layer lining the colon's lumen was completely reconstituted in IL-1α KO mice. In IL-1β KO mice, there were negligible signs of repair. Colon inflammatory manifestations in IL-1 KO mice correlated with secretion of cytokines. Online supplementary figure 1 shows levels of IL-6, a prototypic proinflammatory cytokine, induced mainly by IL-1.
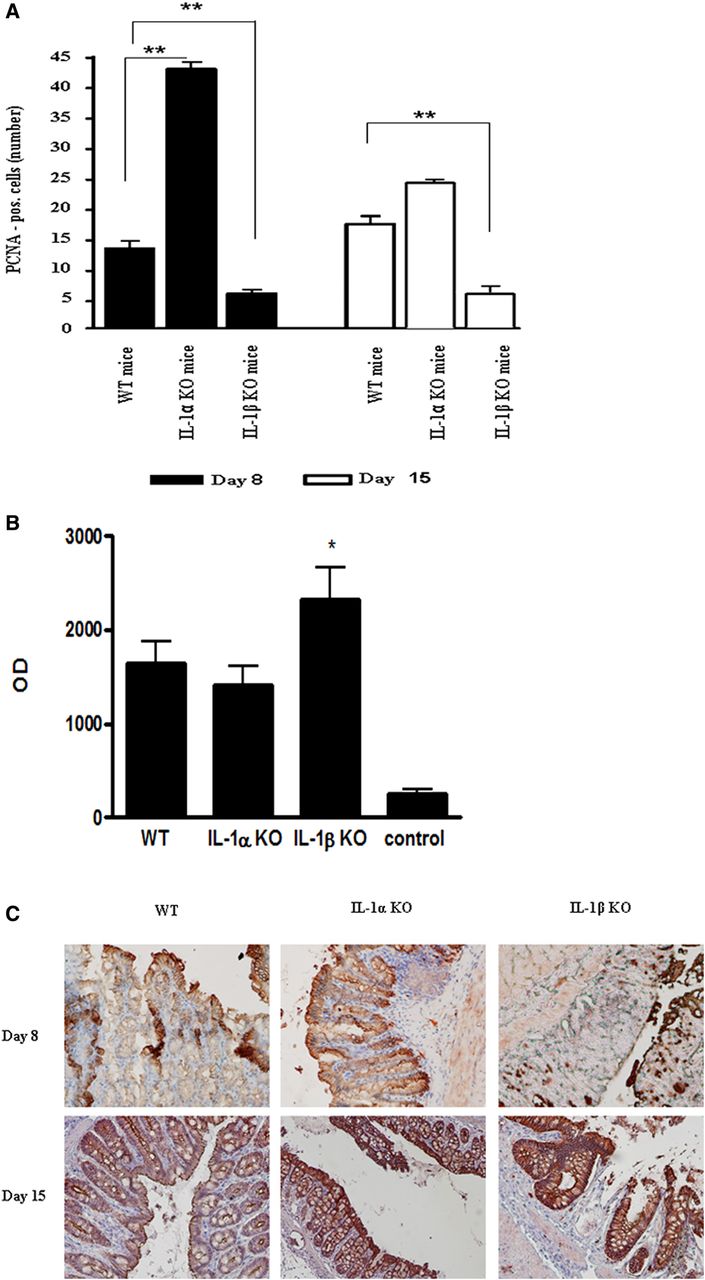
Delayed repair of the epithelial cell barrier in IL-1β deficient mice. (A) Quantification of the number of proliferating cell nuclear antigen-positive cells per crypt on days 8 and 15 after initiation of colitis. The number of positive cells was obtained by counting six random fields per slide (6 mice/group). The data are from three independent experiments, **p<0.001. (B) Plasma levels of FITC-D were measured 2 h after administration of FITC-dextran by spectrophotofluorometry (Safire2, Tecan Group, Männedorf, Switzerland) with an excitation wavelength of 483 nm, and an emission wavelength of 525 nm, using serially diluted samples of the marker as a standard (5–6 mice/group). The data are from three independent experiments, p values were calculated with ANOVA; *p<0.05. (C) Representative immunohistochemical photomicrographs for claudin-3 expression in intestinal crypts on day 8 and 15 after initiation of dextran sodium sulfate treatment (magnification ×400). The picture is from one out of three independent experiments.
The differential expression of IL-1α and IL-1β in colon epithelial and infiltrating leukocytes during acute colitis
As presented in figures 4A, in WT mice treated with DSS, IL-1α was detected at the base of the preserved crypts, mainly in the nuclei of IECs. By contrast, IL-1β was found mostly in inflammatory cells (figure 4B). IL-1α, but not IL-1β, was also detected in IECs under homeostatic conditions in untreated mice (figure 4E,F). In WT and IL-1β KO mice, in areas of severe tissue damage, complete crypt destruction with a loss of cytokeratin-18 expression was observed (figure 4C). In these areas, we detected IL-1α in myeloid infiltrating cells (figure 4D).
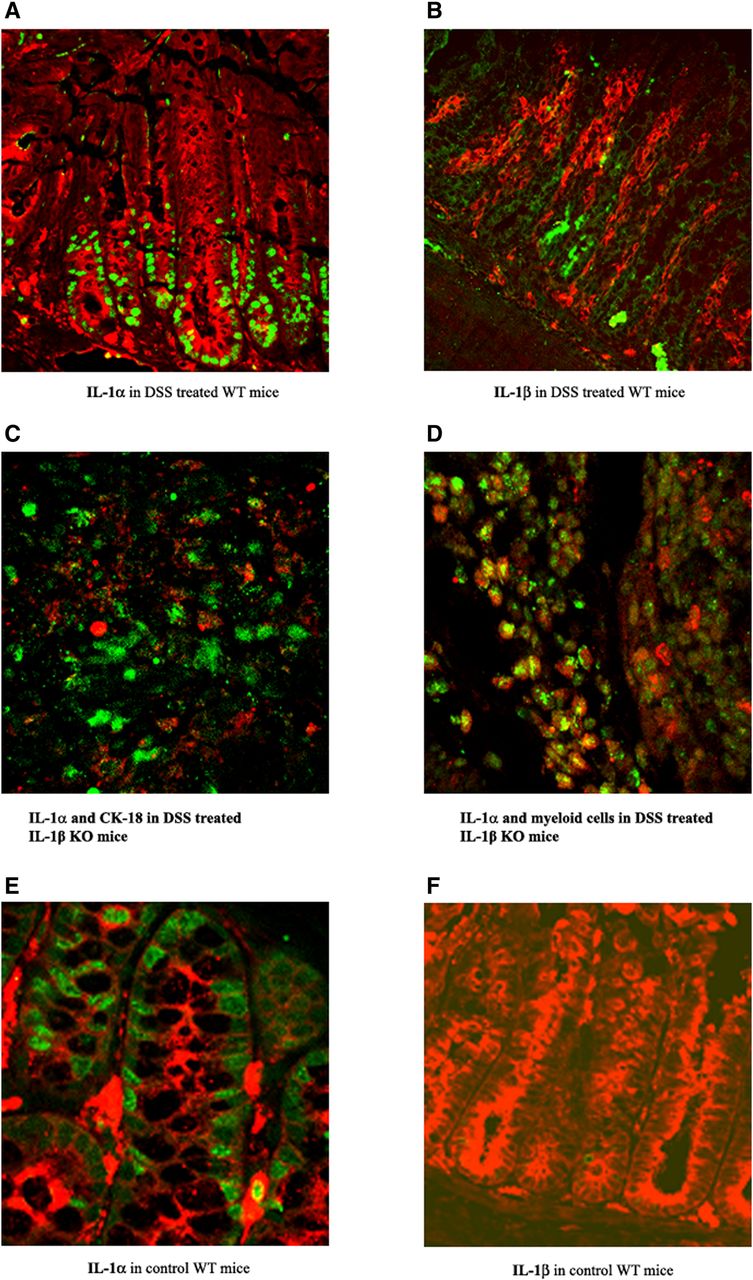
Expression of IL-1α and IL-1β in colons of mice with dextran sodium sulfate-induced colitis. Photomicrographs of colon sections from WT mice stained for: (A) Anti-IL-1α (green) and anticytokeratin-18 (red). (B) Anti-IL-1β (green) and anticytokeratin-18 (CK-18) (red). (C) Colocalisation of IL-1α (green) and anticytokeratin-18 (red) in colon tissue obtained from IL-1β KO mice on day 8. (D) Coexpression of IL-1α and myeloperoxidase-positive cells in colon tissue of IL-1β KO mice on day 8 (magnification ×60). Photomicrographs are from one out of three independent experiments. Representative photomicrographs of colon sections from non-treated (control) mice were stained with antibodies for (E) cytokeratin-18 (red), IL-1α (green, left panel) or (F) IL-1β (green, right panel), (magnification ×60).
IL-1α of epithelial cell origin initiates the inflammatory response and dictates the outcome of acute colitis
Next, we demonstrated the critical role of IEC-derived IL-1α in colon inflammation. We used the Cre-LoxP system to create conditional IL-1α deficient mice, in which IL-1α is specifically deleted in IECs. The absence of IL-1α in IECs was confirmed by quantitative RT-PCR analyses (data not shown). Only mild colitis was observed in IL-1αint/Δ mice, as assessed by weight loss (figure 5A) and tissue damage (figure 5B,C, upper panel). These observations are similar to patterns of colitis in IL-1α KO mice. By contrast, in IL-1αloxP/loxP control mice, a severe form of colitis was observed, similar to that in WT mice.
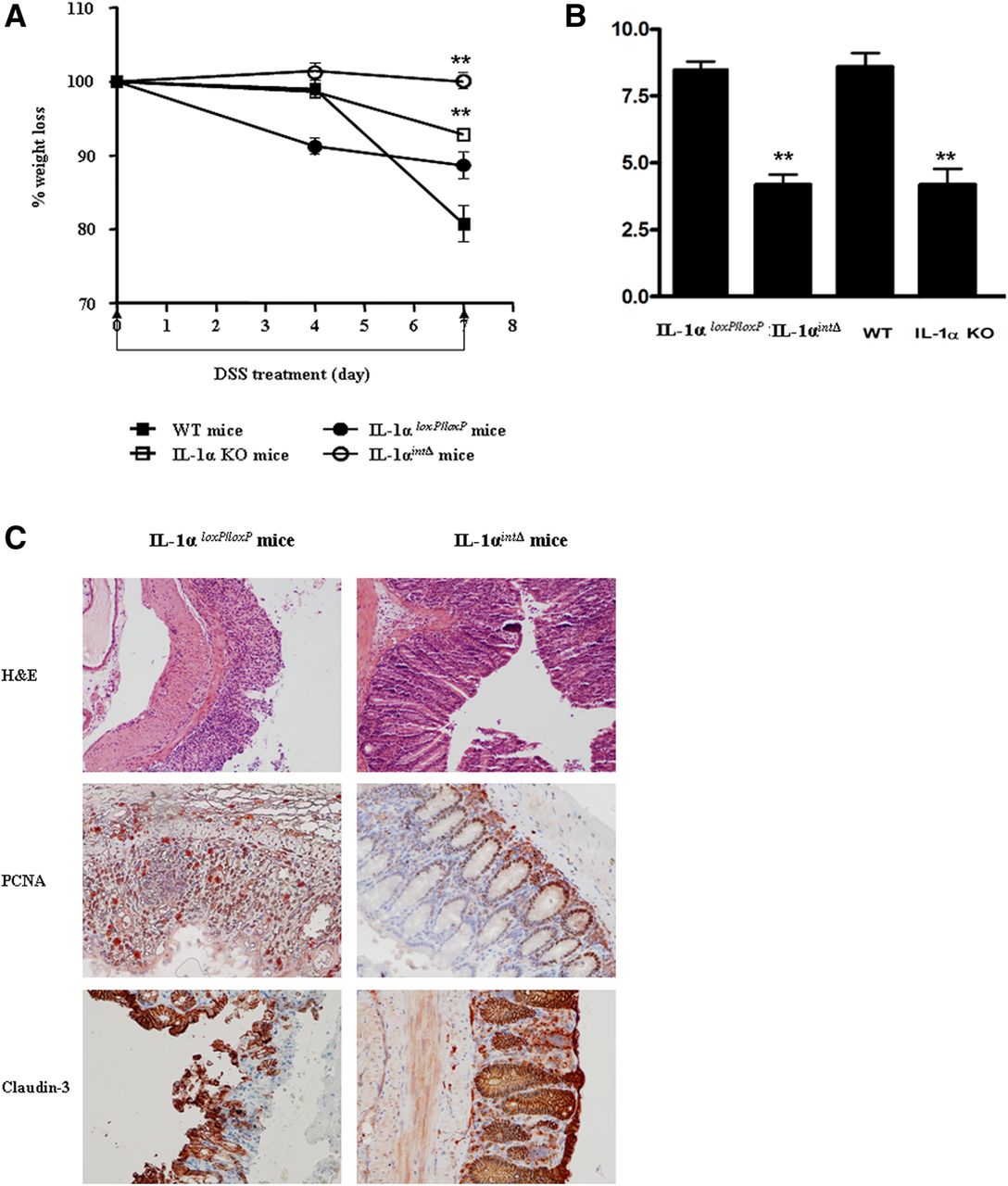
IL-1α from intestinal epithelial cells is the major inducer of inflammation in dextran sodium sulfate (DSS)-induced colitis. (A) Daily changes in body weight in IL-1αintΔ mice and IL-1αloxP/loxP mice and, as controls, in WT and globally IL-1α KO mice (mean±SEM). Body weight changes were calculated by comparing body weight on days 0 and 8, and are expressed as a percentage. The data are representative of two independent experiments, **p<0.001. (B) Total histological score on day 8 after initiation of DSS treatment. Data values are mean±SEM from two independent experiments, **p<0.001. C. Representative photomicrographs obtained from colons on day 8 stained with: H&E (upper panel, magnification ×200), anti-proliferating cell nuclear antigen (middle panel, magnification ×400) and anticlaudin-3 (lower panel, magnification ×200). Photomicrographs are from one experiment of two performed.
Additionally, improved tissue repair was noted in IL-1αint/Δ mice, compared with IL-1αloxP/loxP mice, as assessed by PCNA (figure 5C, middle panel) and claudin-3 (figure 5C, lower panel) stainings. These data clearly indicate that IL-1α of IEC origin plays a dominant role in inducing colon inflammation.
Interactions between IL-1 expressed in non-hematopoietic versus BM-derived cells in acute colitis
Using bone marrow transplantation (BMT), we further assessed the interactions between IL-1 of host-derived non-hematopoietic and BM-derived cells. Patterns of colon damage were determined in mice deficient in IL-1α, IL-1β, or in WT mice that had been lethally irradiated and then reconstituted with BM from WT GFP-transgenic mice. All chimeric mice were assessed for the efficiency of BMT by detection of GFP-positive cells in spleens at the endpoint of experiments (results not shown). In general, colitis was more severe in all groups of chimeric mice and these mice showed an increased mortality rate compared to non-irradiated mice. IL-1β and IL-1α KO mice, after transplantation of WT BM cells, exhibited greater survival rates compared to WT chimeric mice, in which the most severe clinical and histological changes were observed (figure 6A).

The effects of myeloid cell-derived and epithelial cell-derived IL-1α and IL-1β on initiation and repair of colitis. Irradiation chimeras were prepared as indicated in Methods. (A) Survival of WT mice (10–12 mice/group), IL-1α KO (8–10 mice/group) and IL-1β KO (7–10 mice/group) chimeric mice after 7 days of dextran sodium sulfate (DSS) treatment. The data are from two independent experiments. Survival curve was calculated using the Kaplan–Meier method. (B) Representative photomicrographs of H&E stained colon sections from WT and IL-1 KO mice or mice with the indicated bone marrow transplantation treated with DSS (magnification ×200). The pictures are from one of two independent experiments (C). Data values are mean±SEM of histological scores of WT (6 mice/group), IL-1α KO (7 mice/group) and IL-1β KO (6 mice/group) chimeric mice on days 8 and 15, *p<0.05 and **p<0.001. (D) Data values are mean±SEM of proliferating cell nuclear antigen-positive cells in crypts on days 8 and 15. The data are from two independent experiments, **p<0.001. (E) Representative photomicrographs of colons obtained on day 15 and stained with anticlaudin-3 antibodies (magnification ×200). Pictures are from two independent experiments.
Although IL-1α chimeric mice showed intensive tissue damage on day 8, enhanced repair of the epithelium 1 week after cessation of DSS treatment was observed in this group compared to WT mice (figure 6B). In IL-1β chimeric mice, there was less destruction of the epithelium in comparison with other chimeric mice (figure 6B) and non-irradiated IL-1β KO mice treated with DSS (figure 1). The histological score of tissue damage in chimeric mice is shown in figure 6C. Each of these observations suggests that IL-1α from BM-derived cells exacerbates tissue damage, whereas IL-1β from BM-derived cells is important for tissue repair following an initial episode of acute inflammation of the colon.
The data suggest that the total quantity of IL-1 in the relevant compartments, as well as the unique functions of IL-1α and IL-1β in colitis determine the severity of colitis in chimeric mice. This was also confirmed by improved parameters related to tissue repair, such as increased PCNA and claudin-3 expression in colons of IL-1β chimeric mice 1 week after termination of DSS treatment (figure 6D, E, respectively). Reciprocal chimeras, in which WT mice were reconstituted with BM cells from either WT, IL-1α deficient or IL-1β deficient mice, developed severe colitis, with no significant differences between the groups (data not shown).
Improved colon repair in IL-1α deficient mice correlates with increased T cell infiltration
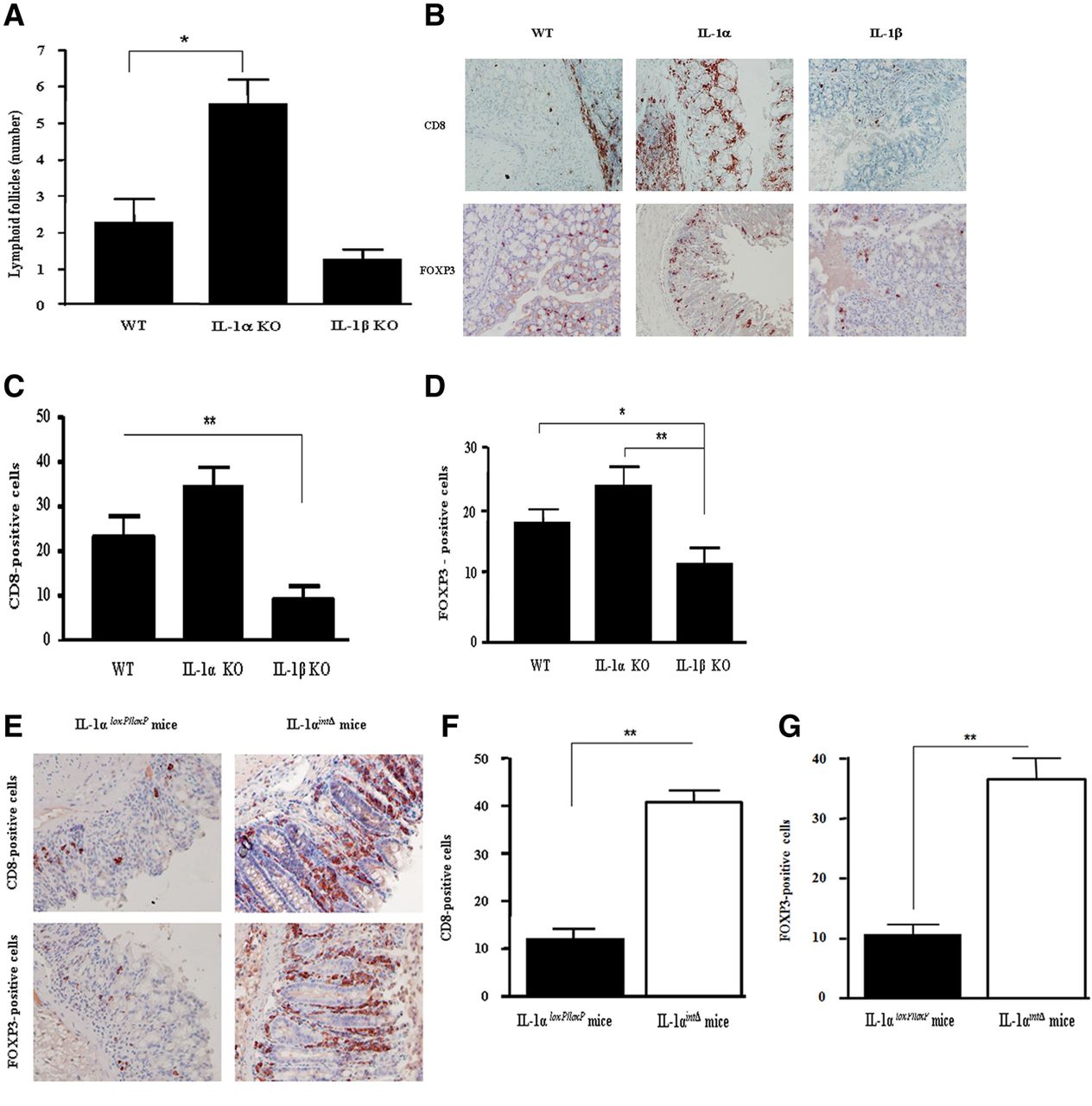
Next, we studied immune mechanisms that might be affected by IL-1α or IL-1β during colon inflammation. In IL-1α deficient mice, which have a mild form of acute DSS colitis compared to WT and IL-1β deficient mice, we observed increased numbers of LF (figure 7A). The increase in the cellular infiltrate in IL-1α deficient mice raises the issue of the composition of this infiltrate. Although there was no significant difference in the number of myeloid cells invading the colon of the three groups of mice during the initial 8 days of acute DSS-induced colitis, an increase in neutrophils in IL-1β KO mice was observed on day 15 (results not shown). Elevation of CD8-positive and Treg cells in colons of DSS-treated IL-1α KO mice was observed compared to WT and IL-1β KO mice (figure 7B–D), with no difference in the number of CD4-positive cells between the three groups. In fluorescence-activated cell sorting analyses of cells obtained from the lamina propria, CD8+ cells were CD3CD25−FOX3−, whereas Tregs were CD3+CD4+CD25+FOX3+ (results not shown).

T cell infiltration in colons of mice with successful repair of dextran sodium sulfate (DSS)-induced colitis. (A) Number of lymphoid follicles in colons after DSS-induced colitis on day 8 (mean±SEM, 8–9 mice/group). Data are from five independent experiments, *p<0.05. (B) Representative photomicrographs of tissue sections from colons on day 8 of DSS treatment, stained with anti-CD8 (upper panel) or anti-FOXP3 antibodies (lower panel), (magnification ×400). Results are from four independent experiments. (C) Mean±SEM number of CD8-positive cells in the same sections as shown in (B), **p<0.001. (D). Mean±SEM number of FOXP3-positive cells calculated from six fields in the same sections as shown in (B), **p<0.001 (E). Colons of control IL-1αloxP/loxP (left panels) and IL-1αintΔ mice (right panels) were stained with anti-CD8 antibodies (upper panel) or anti-FOXP3 antibodies (lower panel). Representative photomicrographs from two independent experiments are shown, (magnification ×400). (F) Mean±SEM number of CD8-positive cells. (G) Mean±SEM number of FOXP3-positive cells. Cell numbers were calculated per six fields, as described in figure 3. The data are from two independent experiments (5–6 mice/group). **p<0.001.
To evaluate the involvement of IEC-derived IL-1α in regulating the T-cell infiltrate in the colon, we used IL-1α int/Δ mice treated with DSS. As in globally IL-1α deficient mice, there was an increase in both CD8−positive T cells and Treg cells in colons obtained on day 8 from DSS-treated IL-1αint/Δ mice (figure 7E–G).
Moreover, we found that transplantation of WT myeloid cells to WT or especially to IL-1α KO mice was accompanied by a reduction in the number of CD8-positive T cells and Treg cells (results not shown). By contrast, BMT of WT cells into IL-1β deficient mice leads to an increase in these populations compared to non-transplanted IL-1β KO mice. Thus, optimal colon repair after acute DSS-induced colitis correlates with recruitment of CD8-positive T cells and Tregs into the colon.
Neutralisation of IL-1α reduces severity of acute colitis
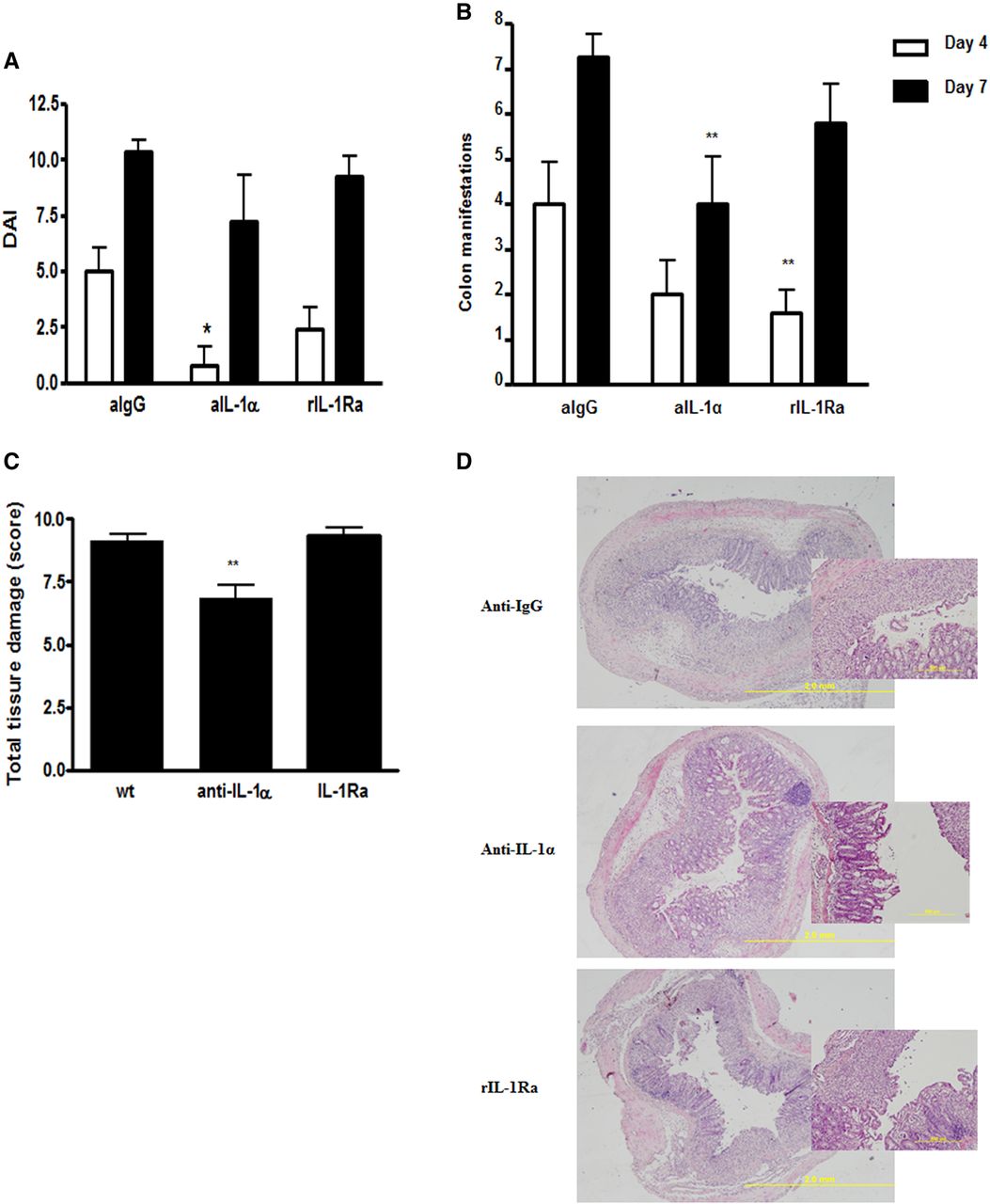
In order to confirm that IL-1α is essential for the inflammatory response in acute colitis, we performed neutralisation experiments with specific anti-IL-1α antibodies, and as a control we used isotype-matched irrelevant antibodies. Mice were also treated with rIL-1Ra, which neutralises both IL-1α and IL-1β. As shown in figure 8A, neutralisation of IL-1α led to a significant decrease in the clinical score on day 4, while on day 7, these differences were not significant. However, colon manifestations and the histological score were significantly decreased in mice treated with anti-IL-1α antibodies (figure 8B,C). Treatment with rIL-1Ra had some effects on day 4, which were not observed at later time intervals. We also assessed the effects of neutralisation of IL-1β by specific antibodies in WT mice and, as expected, there was no significant effect on the DAI or histological score (results not shown), probably due to the severity of the colitis in WT mice, which cannot be further exacerbated.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Neutralisation of IL-1α induces alleviation of acute colitis. (A) Common clinical score (weight loss plus colon manifestations) on days 4 and 7 after initiating dextran sodium sulfate (DSS) treatment. Mice were treated with DSS in the presence of irrelevant (anti-IgG) or anti-IL-1α antibodies, or rIL-1Ra. (B) The score of colon manifestations in mice indicated above is presented (7–9 mice/group). Data represents mean±SEM from two independent experiments. p Values were calculated with ANOVA; *p<0.05. (C) Histological score was calculated in mice treated with DSS together with irrelevant (anti IgG) or anti-IL-1α antibodies, or rIL-1Ra, (6–9 mice/group). Data represents mean±SEM from two independent experiments. p Values were calculated with ANOVA; *p<0.05. (D) Representative photomicrographs of colons of DSS-treated IL-1 deficient and WT mice, sacrificed on day 8 (H&E staining, magnification ×80).
Discussion
In this study, we show non-redundant differential roles for IL-1α and IL-1β in acute DSS colitis; IL-1α is primarily involved in the inflammatory response, whereas, the dominant function of IL-1β is in the repair process of the epithelial barrier of the colon. We believe this is the first study to show evidence that IL-1α, which is readily released from DSS-damaged IECs, is the major mediator of inflammation in DSS colitis by acting initially as an alarmin. In IL-1α deficient mice, inflammation was minimal (figure 1), followed by complete healing (figures 2 and 3). The uniqueness of this finding was validated by conditional deletion of IL-1α in IECs, which showed a remarkably mild and reparative form of colitis (figure 5). Hence, we conclude that IEC-derived IL-1α plays a dominant role in initiation of DSS colonic inflammation. IL-1α released from damaged IECs likely activates inflammatory mediators from colon-resident mononuclear cells, as well as infiltrating cells, which further amplifies the initial inflammatory response.
We, as well as others, have previously reported the alarmin function of cell-associated IL-1α in the initiation of acute inflammation in response to damaged tissue.19 ,21 ,28 ,29 In these studies, the precursor form of IL-1α is constitutively present in the cytosol and/or nucleus of epithelial cells such as keratinocytes.21 We also document constitutive IL-1α in colon IECs (figure 4E). Upon induction of apoptosis, the IL-1α precursor, which bears a nuclear localisation sequence, translocates into the nucleus, where it avidly binds to chromatin, thus preventing the release of precursor IL-1α into the microenvironment.19 However, upon cell necrosis, for example after DSS treatment, the precursor of IL-1α leaves the nucleus, relocates to the cytosol and is released from the cell (figure 4).19 The released IL-1α precursor can be further cleaved in the extracellular microenvironment by neutrophil serine proteases secreted by infiltrating myeloid cells to a more active cytokine that further amplifies colon inflammation.30 Continuous exposure of IECs to the microbiota possibly results in constitutive IL-1α expression, which can induce inflammation upon cell damage. This hypothesis is being investigated in our lab.
Mice deficient in IL-1β develop a severe form of colon inflammation (figure 1), with delayed and poor repair (figures 2 and 3). These observations are consistent with the role of IL-1β in promoting healing and scar formation.17 Indeed, studies with IL-1RI deficient mice show that the atherosclerotic process requires IL-1β to maintain plaque stability and outward vessel remodelling.31 ,32
The contribution of the inflammasome pathway, which leads to processing and secretion of IL-1β and IL-18 in the development of colon inflammation and carcinogenesis has been recently studied (reviewed in refs. 13 33–36 13 ,33–36). In IBD, an interplay between inflammatory pathways in IECs versus myeloid cells controls the outcome of colitis (reviewed in refs. 7 10 12 13 15 34 36 7 ,10 ,12 ,13 ,15 ,34 ,36). Most studies show that in myeloid cells, inflammasome-processed cytokines contribute to inflammation, while in IECs, expression of some of these inflammatory molecules is important for homeostasis and integrity of the colon barrier.12 ,13 ,33–38 Thus, mice deficient in NOD-like receptors (NLRPs), bipartite adaptor protein or caspase-1 are susceptible to DSS-induced colitis and colon carcinogenesis due to inappropriate repair, manifested by impairment of the proliferative capacity of IECs and aberrant reconstitution of the epithelial barrier of the colon.12 ,36–40 Indeed, IL-18 in IECs has been shown to exert a protective role in colon repair.36 ,41 Impaired repair after DSS-induced colitis and enhanced carcinogenesis in Myd88 KO mice was not be observed in IL-1R KO mice, which displayed a similar phenotype to that of WT mice.25 This observation may be explained in light of the data described here, which reveal the opposing effects of the two IL-1 agonistic molecules in colon inflammation. Thus, in IL-1R KO mice, the inflammation-promoting effects of IEC-derived IL-1α and the protective effects of IL-1β are not functional due to the absence of the IL-1R and, therefore, these mice do not display a particular phenotype in DSS colitis. Here, we provide novel data showing that deficiency in IL-1β results in impairment of repair, manifested by increased colon permeability, decreased proliferation of IECs and defects in tight junction proteins (figure 3). Similarly, decreased expression of NLRP3 and diminished IL-1β secretion was recently shown to be associated with increased susceptibility to IBD.42
In BMT experiments, we observed that IL-1β of myeloid cell origin induces repair of DSS-induced tissue damage in mice lacking IL-1β (figure 6). Similarly, in NLRP6 KO mice, the severity of colitis was reversed and repair was induced following transplantation of BM cells from WT mice.43 We observed that reconstitution of IL-1α KO mice with BM cells from WT mice exacerbates colitis. Nevertheless, these mice showed reduced mortality and improved repair of the epithelial barrier (figure 6). In this case, when IL-1β is present in colon non-hematopoeitic cells, additional transplantation of IL-1β of myeloid cell origin possibly exacerbates colon inflammation.
We found a strong correlation between a good repair of acute colitis and the number of intestinal LF (figure 7), which was shown to be important in preserving the epithelial barrier of the colon.44 An abundance of CD8+ and CD4+CD25+FOXP3+ cells was observed in the crypts of mice undergoing good healing of the colon, such as IL-1α KO mice, or mice with conditional depletion of IL-1α in IECs (figure 7). Colon inflammation in DSS-induced colitis is mainly mediated by innate immunity, however, at later stages, adaptive immune cells, mainly T cells, can also be activated to modulate the disease (reviewed in refs. 45 46 45 ,46). Treg cells play an essential role in the maintenance of immune homeostasis and tolerance by suppressing Th1-and Th17-mediated colonic inflammation and were also shown to be capable of reversing established colitis.47 ,48 Additionally, a population of CD8 cells has been described that inhibits IFNγ secretion in CD4 T cells, thereby preventing experimental IBD.49 Further studies on the mechanisms through which the IL-1 agonists affect differentiation and function of lymphocyte populations in the colon are in progress.
Recently, the effector function of IL-1β in activation of Th17 cells in T cell-dependent models of chronic colitis has been shown.50 The goal of further studies will be to elucidate interactions between microenvironment IL-1 and T cell activity in acute colitis.
In conclusion, we found a differential role for IL-1α and IL-1β in colon inflammation and repair. The release of the intracellular precursor of IL-1α from injured IECs initiates colon inflammation and induces recruitment of myeloid cells, which then amplifies the inflammatory response by secreting diverse proinflammatory molecules, including IL-1 agonists. Although IL-1β may contribute to inflammation, the dominant effect of IL-1β in colitis appears to be part of the repair process. In acute colon inflammation, specific neutralisation of IL-1α is beneficial in determining the outcome of the disease, while treatment with rIL-1Ra, which inhibits signalling of both IL-1 molecules, is not effective (figure 8). Elucidating the interactions between the IL-1 molecules expressed in IECs versus myeloid cells in colitis will facilitate the development of therapeutic options, particularly using neutralisation of IL-1α to reduce chronic inflammation and its exacerbation to colon cancer.
Acknowledgments
The authors wish to thank Professor Y Iwakura, Tokyo University, for the IL-1 KO mice, and Professor Yinon Ben-Nerriah for the villin-cre mice. We thank Dr Monica Huszar for her help in the pathology studies. We thank Professor Steffen Jung and Professor Yechiel Zick for their help in the colon permeability assay. Special thanks go to Dr John Simard and Dr Sushma Shivaswamy, XBiotech, USA, for their generous help in the neutralisation experiments. We thank Dr Lyubov Gayvoronsky and Ms Parvin Zarin for their technical assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figure
Footnotes
-
RA and EV contributed equally.
-
Contributors Data acquisition by MB, LL, DF, RMW, SD, PR, IK, TA and EV; pathology support by NZS; study design and paper authorship by MB, CAD, RNA and EV.
-
Funding Ron N Apte was supported by the following grants: FP7: Cancer and Inflammation (INFLA-CARE), Israel Ministry of Science (MOS) jointly with the Deutsches Krebsforschungscentrum (DKFZ), Heidelberg, Germany, the Israel Science Foundation funded by the Israel Academy of Sciences and Humanities, the Israel Cancer Association and the Israel Ministry of Health Chief Scientist's Office. Prof Ron N Apte is an incumbent of the Irving Isaac Sklar Chair in Endocrinology and Cancer, Ben Gurion University of the Negev. Elena Voronov was supported by the FP7: Cancer and Inflammation and the Israel Science Foundation, Israel Cancer Association, the Israel Ministry of Health Chief Scientist's Office and the Israel Science Foundation funded by the Israel Academy of Sciences and Humanities Charles A Dinarello is supported by grants from the National Institutes of Health, AI-15614, AR-45584 and CA-04 6934.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.