Article Text

Abstract
Design Pharmacokinetic and pharmacodynamic parameters of cremophor-paclitaxel, nab-paclitaxel (human-albumin-bound paclitaxel, Abraxane) and a novel mouse-albumin-bound paclitaxel (m-nab-paclitaxel) were evaluated in genetically engineered mouse models (GEMMs) by liquid chromatography-tandem mass spectrometry (LC-MS/MS), histological and biochemical analysis. Preclinical evaluation of m-nab-paclitaxel included assessment by three-dimensional high-resolution ultrasound and molecular analysis in a novel secreted protein acidic and rich in cysteine (SPARC)-deficient GEMM of pancreatic ductal adenocarcinoma (PDA).
Results nab-Paclitaxel exerted its antitumoural effects in a dose-dependent manner and was associated with less toxicity compared with cremophor-paclitaxel. SPARC nullizygosity in a GEMM of PDA, KrasG12D;p53flox/−;p48Cre (KPfC), resulted in desmoplastic ductal pancreas tumours with impaired collagen maturation. Paclitaxel concentrations were significantly decreased in SPARC null plasma samples and tissues when administered as low-dose m-nab-paclitaxel. At the maximally tolerated dose, SPARC deficiency did not affect the intratumoural paclitaxel concentration, stromal deposition and the immediate therapeutic response.
Conclusions nab-Paclitaxel accumulates and acts in a dose-dependent manner. The interaction of plasma SPARC and albumin-bound drugs is observed at low doses of nab-paclitaxel but is saturated at therapeutic doses in murine tumours. Thus, this study provides important information for future preclinical and clinical trials in PDA using nab-paclitaxel in combination with novel experimental and targeted agents.
- Pancreatic Cancer
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Significance of this study
What is already known about this subject
-
Pancreatic ductal adenocarcinoma (PDA) is a stroma-rich tumour that is highly refractory to systemic therapies.
-
nab-paclitaxel (Abraxane) alone and in combination with gemcitabine has recently shown significant clinical activity in human and murine PDA.
-
Secreted protein acidic and rich in cysteine (SPARC) has been proposed as a biomarker for nab-paclitaxel-based chemotherapies by sequestering nab-paclitaxel to concentrate the drug intratumourally and to deplete tumour stroma.
What are the new findings
-
nab-paclitaxel acts in a dose-dependent but SPARC saturable manner in genetically engineered mouse models (GEMM).
-
SPARC ablation in KPfC mice results in desmoplastic ductal pancreas tumours with impaired collagen maturation.
-
A preclinical trial using a novel formulation of mouse-nab-paclitaxel demonstrates robust therapeutic effects independent of SPARC expression.
-
Prolonged treatment with mouse-nab-paclitaxel does not lead to stromal depletion in the KPC mouse model.
How might it impact on clinical practice in the foreseeable future
-
nab-paclitaxel could be investigated with additional stromal depletion agents to maximise treatment effects.
-
Preclinical platforms using mouse-nab-paclitaxel can be employed to identify more powerful combination therapies for early clinical testing.
-
SPARC serum levels should be measured in PDA patients prior to starting nab-paclitaxel-based chemotherapies to determine whether high plasma SPARC levels affect plasma pharmacokinetics of albumin formulated drugs.
Introduction
Pancreatic ductal adenocarcinoma (PDA) is considered one of the most aggressive and lethal human malignancies with increasing incidence and a 5-year survival rate of less than 5%.1 Over the past decades, intensive efforts to develop novel targeted therapies in combination with various chemotherapies have not yielded a significant breakthrough, and the standard of care chemotherapy has remained gemcitabine despite only marginal effects on patient survival.2 Oxaliplatin, irinotecan, leucovorin and 5-fluorouracil (FOLFIRINOX) has recently been reported to achieve a significant survival benefit for patients with metastatic PDA compared with gemcitabine (11.1 vs 6.8 months); however, increased rates of toxicity will likely limit the frequent clinical use of this regimen in PDA patients.3 Histologically, PDA is characterised by abundant tumour stroma with activated cancer-associated fibroblasts (CAFs) and immune cells that contribute to a dense and highly dynamic tumour microenvironment (TME) around neoplastic ductal cells.4 The distinct stromal architecture has increasingly been appreciated to create physical barriers for drug delivery and also to provide additional biochemical signals that collectively promote the resistance of PDA cells to systemic and targeted therapies.5–13 Secreted protein acidic and rich in cysteine (SPARC/osteonectin/BM40) is an albumin-binding 42-kDa matricellular glycoprotein that has recently gained significant clinical interest as a potential biomarker in PDA. Indeed, SPARC is overexpressed by fibroblasts in the TME of human PDA and has been shown to inversely correlate with survival.14 ,15 A novel drug formulation of paclitaxel bound to albumin (nab-paclitaxel, Abraxane) has been hypothesised to accumulate in and potentially deplete PDA tumour stroma via binding of albumin to SPARC.16 A phase I/II trial of gemcitabine in combination with nab-paclitaxel showed promising overall survival rates and suggested the potential usefulness of SPARC as a predictive biomarker.17 More recently, results from the first randomised phase III study of nab-paclitaxel and gemcitabine Metastatic Pancreatic Adenocarcinoma Clinical Trial, MPACT) have confirmed initial observations of clinical benefit and showed a significant median survival improvement over gemcitabine monotherapy (8.5 vs 6.7 months) in patients with metastatic PDA.18 In parallel, our laboratory has confirmed the antitumour efficacy of nab-paclitaxel and gemcitabine and discovered a synergistic drug–drug interaction where nab-paclitaxel impaired gemcitabine metabolism due to reactive oxygen species (ROS)-mediated degradation of cytidine deaminase.19 Following this encouraging data, nab-paclitaxel plus gemcitabine is currently being evaluated for registration as novel first-line treatment for PDA patients. Furthermore, a recent phase II study found preliminary activity of nab-paclitaxel monotherapy in patients who progressed on gemcitabine-based therapy.20
The use of biomarkers to select effective targeted and cytotoxic therapies and predict response to treatment for individuals (‘personalised medicine’) is becoming a reality in certain types of cancer but has remained elusive in PDA. Recent clinical data suggested that expression of SPARC in the stroma of archived tumour tissue could predict for response to nab-paclitaxel and gemcitabine,17 and a detailed analysis of tumour specimen from the MPACT study is still pending. Preclinical and clinical data on the potential of stromal SPARC to determine treatment response to nab-paclitaxel, sequester nab-paclitaxel to concentrate the drug intratumourally, and to deplete tumour stroma have been conflicting in different tumour entities and hampered by methodological differences.20–23 A preclinical study in patient-derived xenografts reported stromal depletion upon nab-paclitaxel treatment,17 whereas our own results in a genetically engineered mouse model (GEMM) of PDA did not indicate such effects following short-term treatment with nab-paclitaxel.19 A limitation of our prior study was anaphylaxis due to the use of human-albumin-bound paclitaxel, and thus, did not allow us to investigate longer term treatment effects such as stromal depletion.19 Furthermore, the interaction of murine SPARC and human albumin does not recapitulate the genuine biological situation and may possibly bias the results.
Here, we determine the pharmacokinetic and pharmacodynamic profile of nab-paclitaxel in murine PDA in vivo and assess the preclinical effectiveness in GEMMs of PDA in a SPARC-dependent manner by using a novel experimental mouse-albumin paclitaxel (m-nab-paclitaxel) that was specifically formulated for this purpose. Thus, our study provides important information for future preclinical and clinical trials in PDA using nab-paclitaxel in combination with novel experimental and targeted agents.
Materials and methods
Additional details are provided in online supplementary materials and methods.
Genetically engineered mice
The following genetically engineered mice were used for this study: SPARC−/− mice (B6;129S-Sparctm1Hwe/J) were purchased from Charles River (Margate, UK).24 LSL-KrasG12D/+;LSL-Trp53R172H/+;Pdx-1-Cre (KPC) and KrasG12D/+;Trp53flox/−;p48-Cre (KPfC) mice were used for the experiments, and both models develop advanced and metastatic PDA with 100% penetrance at an early age recapitulating the full spectrum of histopathological and clinical features of human PDA.25 Mice were housed at a 12-h light, 12-h dark cycle. All procedures were conducted in accordance with the institutional and national guidelines.
Drugs
nab-paclitaxel (Abraxane) and mouse (m)-nab-paclitaxel were formulated and provided by Celgene Corporation (USA) and resuspended in sterile normal saline at 24 mg/mL, 12 mg/mL or 6 mg/mL, and intravenously administered at 120 mg/kg, 60 mg/kg or 30 mg/kg. Paclitaxel was provided by Addenbrooke's Hospital Pharmacy (Cambridge, UK), resuspended in Cremophor at 6 mg/mL or 2 mg/mL and administered at 10 mg/kg or 30 mg/kg. The amount of paclitaxel is equal in 30 mg/kg nab-paclitaxel and 30 mg/kg cremophor-paclitaxel; 60 mg/kg nab-paclitaxel in mice corresponds to approximately 180 mg/m2 nab-paclitaxel in humans.26
LC-MS/MS of paclitaxel
Fresh frozen tumour samples from different time points were processed and analysed for paclitaxel concentrations using liquid chromatography tandem mass spectrometry (LC-MS/MS). Briefly, samples were extracted with 100% acetonitrile containing 2H5-paclitaxel as internal standard, and chromatography was done on an acquity ultra performance liquid chromatography (UPLC), high strength silica (HSS) T3 50 mm×2.1 mm id., 1.8 µm column (Waters, Hertfordshire, UK). The mobile phase was (A) 0.1% acetic acid: (acetonitrile:methanol (1:1)) 70:30 and (B) 0.1% acetic acid: (acetonitrile:methanol (1:1)) 10:90. The gradient, at a flow rate of 0.3 mL/min, was 100% A for 0.6 min changed to over 0.2 min to 100% B and maintained for 2 min, changed back to 100% A over 0.2 min and held for 1.5 min to give a total run time of 4.5 min. LC-MS/MS was performed on a triple stage quadrupole (TSQ) Vantage mass spectrometer (Thermo Scientific, Waltham, MA) fitted with a heated electrospray ionisation (HESI-II) probe operated in positive mode at a spray voltage of 5 KV and capillary temperature of 350°C. Quantitation was done by multiple reaction monitoring of the transitions 876.4–308.1 and 881.4–308.1 for paclitaxel and the internal standard, respectively, and data acquisition was done using LC Quan2.5.6 (Thermo Fisher Scientific, Waltham, MA).
Therapeutic intervention studies with m-nab-paclitaxel
Following weekly manual palpation from 2 months of age, KPC and KPfC mice were subjected to high-contrast ultrasound screening using the Vevo 2100 System with a MS250, 13–24 MHz scanhead (Visual Sonics, Inc, Amsterdam, NL). Mice with tumour diameters of 5–9 mm were randomised and enrolled into the therapeutic intervention studies: m-nab-paclitaxel was intravenously administered at 60 mg/kg in tumour-bearing mice according to the outlined treatment schedules. During the study, tumour growth was quantified on day 3 and day 7 by measuring tumour volumes using reconstructed three-dimensional (3D) ultrasonography with the integrated Vevo 2100 software package. The last administration of m-nab-paclitaxel was given 2 h prior to endpoint.
Western blot analysis
Western blots were performed as previously described.27 The following primary antibodies were used: Hsp90 (Cell Signaling) and murine SPARC (R&D Systems). Membranes were incubated with secondary HRP-antibodies (Jackson ImmunoResearch) and developed using the enhanced chemiluminescence (ECL) detection system (GE Healthcare).
Histological examination
Tissues were fixed in 10% neutral buffered formalin for 24 h and transferred to 70% ethanol. Tissues were embedded in paraffin, and 3–5 µm sections were processed for H&E staining, Herovici (Sigma) and immunohistochemistry using standard protocols as previously described.10 The following antibodies were used: murine SPARC (R&D Systems, AF942), Cleaved Caspase-3 (Cell Signaling Technology, 9661), CD31 (BD Pharmingen, 553370), Ki-67 (Dako, M7249) and phospho-histone-3 (Upstate, 06-570). Images were acquired on an Olympus BX51 microscope (Olympus) or Aperio XT automated scanning system and Imagescope 10 software (Leica). More information can be found in online supplementary material and methods.
Statistical analysis
Statistical analysis was carried out using GraphPad Prism V.5.01 (GraphPad Software). The Mann–Whitney non-parametric t test was used and results are presented as mean±SE. p<0.05 was considered to be significant.
Results
Pharmacokinetics and pharmacodynamics of cremophor-paclitaxel and nab-paclitaxel in KPC tumours
SPARC is overexpressed in the pancreatic tumour microenvironment in KPC mice,19 predominantly by cancer-associated fibroblasts, and to a lesser extent in tumour cell lines (see online supplementary figure 1A). Since this expression pattern is similar to that described with PDA patients,14 ,15 this mouse model represents a tractable experimental platform to interrogate the interaction of SPARC and nab-paclitaxel.
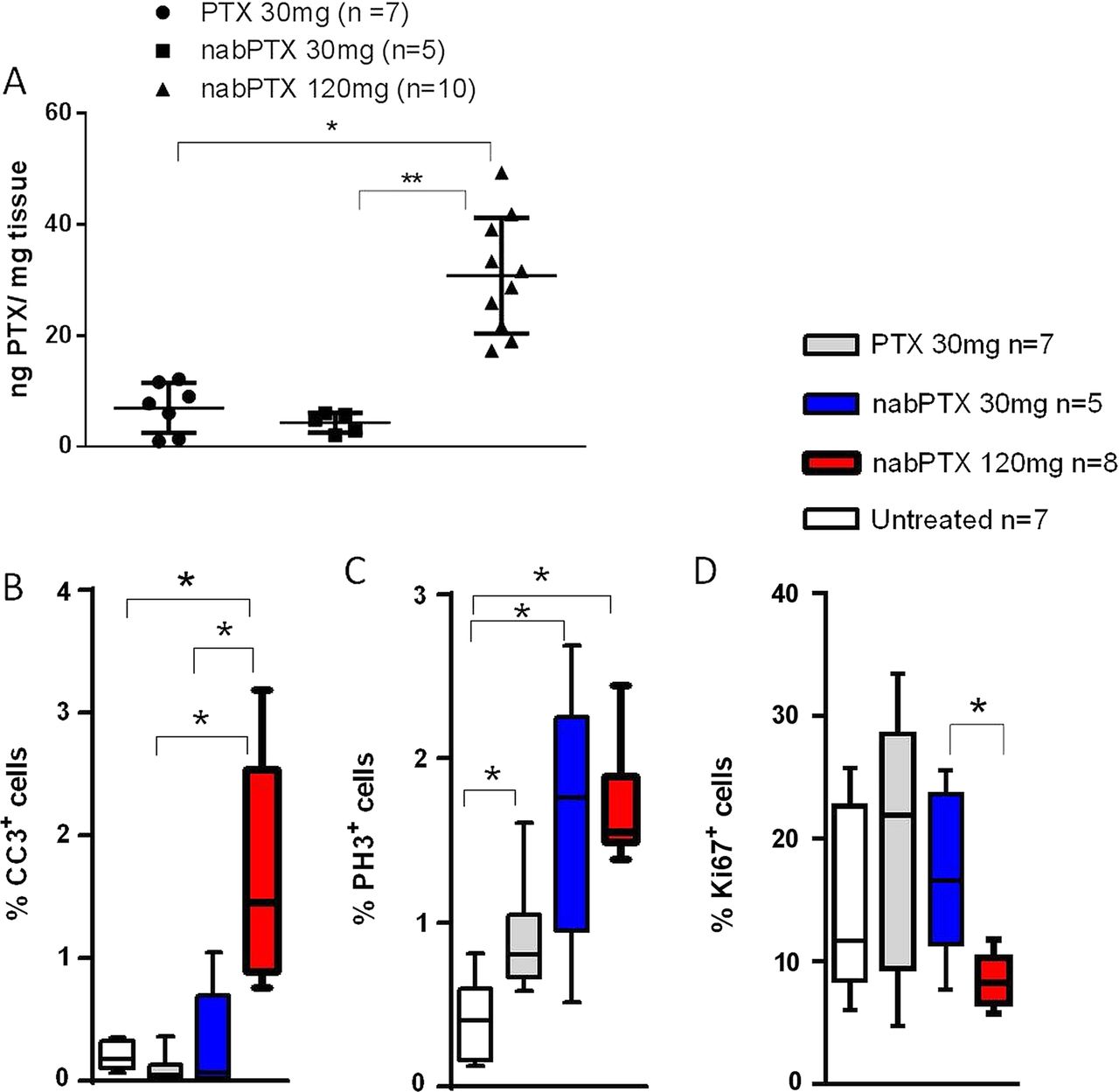
To systematically compare pharmacokinetics and pharmacodynamics of nab-paclitaxel and cremophor-paclitaxel, tumour-bearing KPC mice were intravenously administered a single dose of equimolar (30 mg/kg) cremophor-paclitaxel (n=12), nab-paclitaxel (n=5) and equitoxic 120 mg/kg nab-paclitaxel (n=10). Notably, 5/12 mice (43%) experienced acute toxicity-related health issues with physical inactivity, loss of body temperature and respiratory distress following the administration of 30 mg/kg cremophor-paclitaxel, and those mice had to be culled prior to the defined endpoint. In contrast, 120 mg/kg nab-paclitaxel was well tolerated in all 10 mice treated. Tissues were collected 4 h postdose and assessed for intratumoural paclitaxel concentrations by LC-MS/MS and pharmacodynamic parameters by immunohistochemistry. Strikingly, equimolar doses of cremophor-paclitaxel and nab-paclitaxel (30 mg/kg) resulted in similar intratumoural paclitaxel concentrations (mean: 6.9 ng/mg, SD:±4.5 vs 4.3 ng/mg±1.75, p=0.28, figure 1A), suggesting that intratumoural paclitaxel delivery was independent of the albumin nanoformulation. The maximum tolerated dose (MTD) of 120 mg/kg nab-paclitaxel resulted in approximately fourfold higher paclitaxel levels (30.8 ng/mg±10.4, figure 1A), indicating a nearly linear intratumoural accumulation of nab-paclitaxel with significantly less acute systemic toxicity compared with 30 mg/kg cremophor–paclitaxel.

Single-dose pharmacokinetic and pharmacodynamic analysis of cremophor-paclitaxel and nab-paclitaxel in KPC tumours (A) Tumour-bearing KPC mice were treated with a single dose of cremophor-paclitaxel (30 mg/kg), nab-paclitaxel at 30 mg/kg or nab-paclitaxel at 120 mg/kg. Tumour tissues were harvested and assessed for intratumoural paclitaxel levels 4 h later by liquid chromatography tandem mass spectrometry (LC-MS/MS) (n≥5; *p=0.001; ** p<0.001). (B) Automated quantification of 30 randomly chosen fields for cleaved caspase-3 (CC3, *p<0.01), (C) phospho-histone H3 (*p<0.02) and (D) ki67 proliferation (*p<0.02) immunohistochemistry. Cremophor-paclitaxel LC-MS/MS results from figure 1A have been previously published by Frese et al.19
Paclitaxel has been shown to elicit its antitumoural effects through induction of apoptosis and cell cycle arrest in G2-M. We therefore assessed the effects of the different dosing regimens in KPC tumours by immunohistochemistry for proliferation marker ki-67, apoptosis marker cleaved caspase 3 (CC3) and phosphorylated histone H3 (pH3) as a marker for aberrant mitotic figures. At the MTD of 120 mg/kg, nab-paclitaxel induced significant levels of apoptosis compared with untreated (p<0.001), cremophor-paclitaxel (p<0.001) and low-dose nab-paclitaxel (p<0.01) treated tumours (figure 1B). Notably, cremophor-paclitaxel and low-dose nab-paclitaxel had no significant effects on tumour cell death compared with untreated controls. The appearance of aberrant mitotic figures that contained an abundance of phosphorylated histone H3 was increased following treatment with cremophor-paclitaxel, low-dose nab-paclitaxel and most significantly, in the 120 mg/kg nab-paclitaxel cohort (figure 1C, p<0.001). Again, equimolar doses of nab-paclitaxel and cremophor-paclitaxel did not show significant differences in the percentage of pH3-positive cells (p=0.15, figure 1C). nab-Paclitaxel and cremophor-paclitaxel decreased overall proliferation rate of KPC tumours marginally, and although we found a significant difference in tumour proliferation between low-dose and high-dose nab-paclitaxel (p<0.02), comparison against untreated KPC tumours failed to reach significance (figure 1D; p=0.09).
Overall, these results suggest that nab-paclitaxel and cremophor-paclitaxel have comparable pharmacokinetic and pharmacodynamic properties in murine pancreas tumours with endogenous SPARC expression. However, nab-paclitaxel can be dosed at least four times higher than cremophor-paclitaxel due to improved tolerability and therefore exerts significantly better antitumoural effects in vivo.
m-nab-Paclitaxel extended intervention study in the KPC mouse model
nab-Paclitaxel is formulated with human albumin and thus causes fatal anaphylactic reactions in mice following repeated intravenous injections.19 Therefore, mechanisms such as stromal depletion that require longer term follow-up and repeated injections could not be properly investigated in the past. In order to circumvent these issues, we preclinically assessed a novel, mouse albumin-bound paclitaxel that was specifically formulated for experimental purposes and termed murine (m)-nab-paclitaxel. First, we compared the bioavailability of 30 mg/kg nab-paclitaxel (n=5) and 30 mg/kg m-nab-paclitaxel (n=5) in healthy C57B6 wild-type mice and demonstrated that plasma and tissue concentrations of paclitaxel were comparable between the two compounds following i.v. administration (see online supplementary figure 2A,B).

m-nab-Paclitaxel extended (five doses) intervention study in the KPC model. (A) Automated quantification of 30 randomly chosen fields for phospho-histone H3 and (B) cleaved caspase-3 immunohistochemistry (*p<0.001) following treatment with 60 mg/kg every 3 days for five doses (q3d×5) m-nab-paclitaxel and compared with untreated (UNtx) KPC tumours. (C) Representative H&E stainings from untreated versus long-term m-nab-paclitaxel-treated KPC tumours show no relevant stromal collapse or differences in secreted protein acidic and rich in cysteine expression.
We then determined that 60 mg/kg q3d m-nab-paclitaxel was the MTD in wild-type mice when weight loss became apparent on day 15 (data not shown). To assess whether this dose of m-nab-paclitaxel depletes tumour stroma in KPC mice, tumour-bearing mice (tumour diameter 5–9 mm by 3D ultrasound) were identified and treated over the course of 15 days (see online supplementary figure 2C). Immunohistochemical analyses demonstrated that treatment with m-nab-paclitaxel significantly increased the number of aberrant mitotic figures that contained an abundance of phosphorylated histone H3 compared with untreated KPC tumours (figure 2A, p<0.001). Furthermore, m-nab-paclitaxel significantly increased the number of apoptotic neoplastic cells while sparing stromal fibroblasts in the tumour (figure 2B, p<0.001). Histological examination of the tumour stroma showed that there was no difference in stromal deposition or SPARC expression following m-nab-paclitaxel administration (figure 2C). Although we cannot exclude the possibility that further prolongation of treatment may affect the stromal compartment, it is worth noting that stromal depletion in patient-derived xenografts required only five doses of nab-paclitaxel.17 Taken together, our results suggest that extended treatment with 60 mg/kg m-nab-paclitaxel is well tolerated for 15 days and elicits substantial neoplastic cell death while sparing the stromal compartment in KPC tumours.
SPARC-dependent pharmacokinetics of m-nab-paclitaxel
To investigate the potential interaction between the albumin-binding protein SPARC and m-nab-paclitaxel in SPARC-deficient mice, a pharmacokinetic tissue uptake study was undertaken. In C57B6 wild-type mice, SPARC can be predominantly detected in plasma (n=7) and testis tissue (n=2), whereas kidney (n=2), liver (n=2) and normal pancreas tissues (n=2) show very low or undetectable levels of SPARC protein (see online supplementary figure 3A,B). Paclitaxel concentrations were measured in plasma, kidney and testis samples at seven different time points (0.5 h, 1 h, 2 h, 4 h, 8 h, 20 h and 48 h) in SPARC+/+ (n≥3 mice per time point, n=30 in total) and SPARC−/− mice (n≥3 mice per time point, n=31) following a single intravenous injection of 30 mg/kg m-nab-paclitaxel. Strikingly, the initial phase of paclitaxel distribution from the blood circulation to peripheral compartments (alpha phase) was delayed in SPARC+/+ mice compared with SPARC−/− mice (figure 3A). Plasma paclitaxel levels were significantly increased after 1 h (p<0.01) and 2 h (p<0.02) postinjection of m-nab-paclitaxel, and plasma paclitaxel area under the curve (AUC) was increased by 61.7% in SPARC+/+ versus SPARC−/− (24 100 ng h/mL vs 14 900 ng h/mL) (figure 3A). Accordingly, paclitaxel concentrations were also found to be increased in SPARC+/+ kidney and testis tissue at 1 h (p<0.01) and 2 h (p<0.005) compared with SPARC−/−. The AUC was increased by 18.3% (218.4 vs 184.5 ng h/mL) for kidney, and only marginally increased by 2.7% (70.8 vs 68.9 ng h/mL) in testis tissue (figure 3B,C). Notably, the paclitaxel AUC increase in kidney and testis did not correlate with SPARC expression as SPARC is only expressed in testis but not in kidneys of SPARC+/+ mice (see online supplementary figure 3A). Importantly, SPARC expression in testis tissue did not affect the mean vessel density (see online supplementary figure 3C), and therefore cannot explain the increased AUC. Since kidney and testis samples were not terminally perfused with saline prior to analysis, we hypothesise that increased paclitaxel concentrations in SPARC+/+ organs are caused by higher quantities of blood-borne paclitaxel.

Secreted protein acidic and rich in cysteine (SPARC)-dependent pharmacokinetics of m-nab-paclitaxel. (A–C) Cohorts of SPARC−/− and SPARC+/+ mice were treated with a single dose of 30 mg/kg m-nab-paclitaxel, and plasma, kidney and testis samples were harvested at seven different time points (0.5 h, 1 h, 2 h, 4 h, 8 h, 20 h and 48 h) in SPARC+/+ (n≥3 mice per time point, n=30 in total) and SPARC−/− mice (n≥3 mice per time point, n=31) and analysed by liquid chromatography tandem mass spectrometry (LC-MS/MS) (*p<0.02). (D) Cohorts of SPARC−/− (n=20) and SPARC+/+ (n=20) mice were treated with 10 mg/kg cremophor-paclitaxel and plasma samples were analysed by LC-MS/MS for paclitaxel at four different time points (0.5 h; 1 h; 2 h and 4 h; n=5 mice for each time point).
To investigate whether SPARC deficiency accounts for differences in paclitaxel clearance, we treated SPARC+/+ (n=20) and SPARC−/− mice (n=20) with low-dose cremophor-paclitaxel (10 mg/kg) at four different time points (0.5 h, 1 h, 2 h and 4 h, n=5 per time point) and detected identical plasma paclitaxel concentrations (figure 3D), suggesting that the increased levels of nab-paclitaxel do not stem from altered paclitaxel metabolism but are due to a specific interaction between the albumin formulation of paclitaxel and circulating or endothelial SPARC.
SPARC-deficient GEMM of PDA (KPfC SPARC−/−) develop pancreas tumours with impaired collagen maturation
Since SPARC has been hypothesised to sequester albumin-bound drugs such as nab-paclitaxel and thereby concentrate the drug intratumourally, we sought to generate a GEMM with genetically ablated SPARC to provide an experimental platform for preclinical investigations.
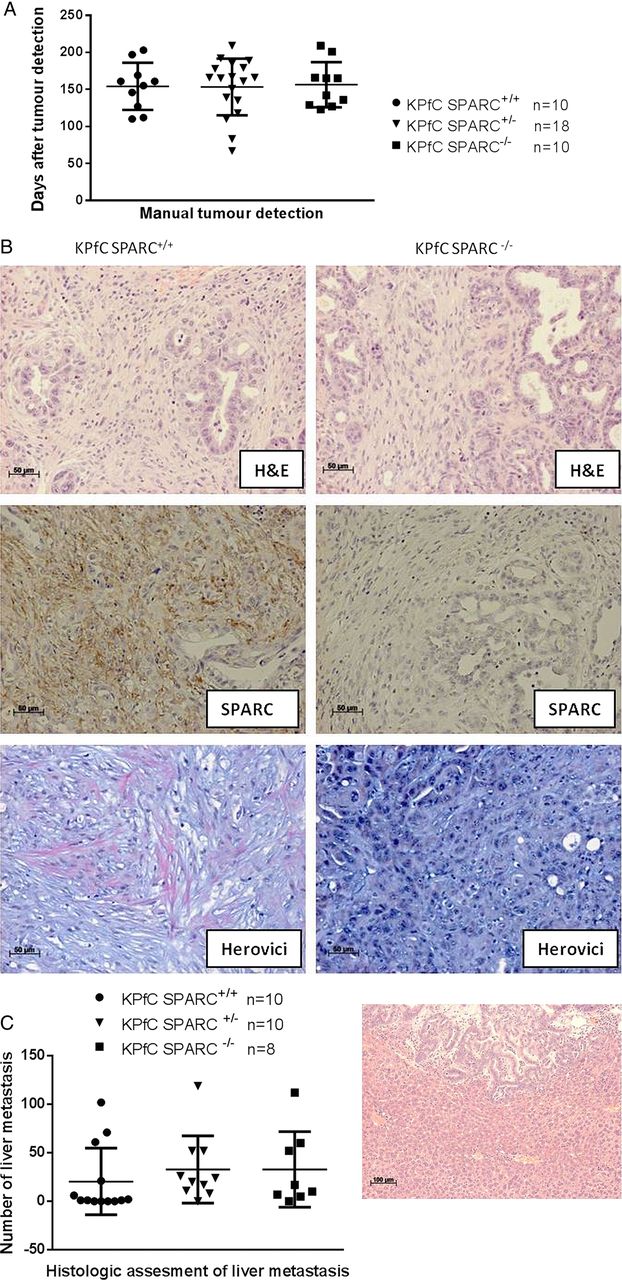
KrasLSL-G12D;p53flox/+;p48Cre (KPfC) mice were generated and developed metastatic PDA with a latency and pathophysiological characteristics indistinguishable from the classic KPC model. The KPfC mice were interbred with SPARC−/− mice to generate cohorts that were SPARC+/+ (n=10), SPARC+/− (n=18) or SPARC−/− (n=10) and monitored for pancreatic tumour development from 2 months of age by weekly manual palpation. SPARC expression did not affect tumour incidence and latency, and pancreatic tumour onset was comparable among the cohorts (SPARC+/+, mean: 154 days; SPARC+/−, 153 days; SPARC−/− 156 days; range 67–209 days, figure 4A). Also, genetic SPARC ablation did not affect the histological appearance of tumours and resulted in murine ductal adenocarcinoma with desmoplastic features indistinguishable from the traditional KPC model (figure 4B). Overall proliferation rate and mean vessel density as assessed by Ki67 and CD31 immunohistochemistry did not show significant differences dependent on the SPARC status (see online supplementary figure 4A,B). Interestingly, detailed histological analysis of the extracellular matrix composition revealed impaired collagen maturation in KPfC SPARC−/− mice as evidenced by Herovici stains (figure 4B), supporting the role of SPARC in collagen production and turnover.28 Following pancreatic tumour development, systemic disease such as haemorrhagic ascites, cachexia and widespread liver metastases was observed in the majority of mice. At endpoint, the metastatic tumour burden in the liver did not significantly differ depending on the SPARC status (figure 4C).

Secreted protein acidic and rich in cysteine (SPARC) deficient genetically engineered mouse model (GEMM) of pancreatic ductal adenocarcinoma develop pancreas tumours with impaired collagen maturation. (A) Tumour incidence following weekly manual palpation of KPfC mice from 2 months of age. (B) Representative H&E from KPfC SPARC+/+ and KPfC SPARC−/−mice shows desmoplastic ducal adenocarcinoma (upper panel). Immunohistochemistry for SPARC confirms genetic ablation of SPARC (middle panel), and Herovici stains show decrease in mature collagen fibres (pink) in KPfC SPARC−/− mice (lower panel). (C) Quantification of five serial liver sections (100 µm apart) shows no significant difference in metastatic burden upon SPARC heterozygous or homozygous deletion in KPfC mice.
SPARC-dependent pharmacokinetics and pharmacodynamics of m-nab-paclitaxel in the KPfC mouse model
To test whether the intratumoural accumulation of m-nab-paclitaxel is dependent upon the expression of SPARC, we administered m-nab-paclitaxel at therapeutic doses (60 mg/kg) to KPfC SPARC−/− (n=11) and KPfC SPARC+/+ (n=8) mice with established tumours of comparable size. Paclitaxel concentrations were measured in tumour biopsies, plasma and kidney samples. Surprisingly, paclitaxel concentrations did not significantly differ between SPARC+/+ and SPARC−/− pancreatic tumours, plasma and kidney samples 2 h after dosing (figure 5A, see online supplementary figure 5A,B), indicating that neither circulating nor tumoural SPARC sequesters nab-paclitaxel to accumulate the drug intratumourally during this timeframe. Furthermore, we conducted a short-term intervention study that compared the tumour growth in KPfC SPARC+/+ (n=6) and KPfC SPARC−/− mice (n=5) with 60 mg/kg m-nab-paclitaxel dosed 3× over 7 days (figure 5B, see online supplementary figure 5C,D). Final tumour volumes in KPfC SPARC+/+ versus KPfC SPARC−/− mice did not significantly differ from each other following 7 days of 60 mg/kg m-nab-paclitaxel treatment (mean: 152 mm3±43 vs 193 mm3±27; figure 5C). In line with treatment responses observed by 3D ultrasound, levels of intratumoural apoptosis were not significantly different in KPfC SPARC+/+ and KPfC SPARC−/− pancreatic tumours (figure 5D). Furthermore, weight loss (not shown) or hematological toxicity was not affected by the SPARC status upon m-nab-paclitaxel treatment (see online supplementary figure 5E). In summary, our data suggest that although m-nab-paclitaxel exerts significant antitumour effects at 60 mg/kg by induction of apoptotic cell death, drug delivery and response to treatment are independent of SPARC expression.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Pharmacokinetics and pharmacodynamics of m-nab-paclitaxel in the KPfC secreted protein acidic and rich in cysteine (SPARC) mouse model. (A) Cohorts of KPfC SPARC+/+ (n=8) and KPC SPARC−/− mice (n=11) were treated with 60 mg/kg m-nab-paclitaxel, and tumour samples were taken 2 h after dosing and analysed by liquid chromatography tandem mass spectrometry (LC-MS/MS). (B) Representative high-resolution ultrasound picture of typical KPfC murine pancreatic tumour. (C) Quantification of tumour volume growth using biweekly three-dimensional high-resolution ultrasound shows no significant decrease in tumour burden on day 7 in KPfC SPARC+/+ versus KPfC SPARC−/− pancreatic tumours following three injections of 60 mg/kg m-nab-paclitaxel over 7 days. (D) Computer-based quantification of apoptosis (cleaved caspase-3) in pancreatic tumours from KPfC SPARC+/+ (n=6) and KPfC SPARC−/− mice (n=5) treated with 60 mg/kg m-nab-paclitaxel. All animals were sacrificed 2 h after the last dose of m-nab-paclitaxel.
Discussion
Nanoparticle albumin-bound (nab)-paclitaxel, an albumin-bound-stabilised paclitaxel formulation, has recently demonstrated significant improvements in median survival of patients with metastatic PDA when combined with gemcitabine.18 Since this drug combination represents the first therapeutic regimen that significantly extends survival of advanced stage PDA patients and is accompanied by an acceptable toxicity profile, nab-paclitaxel and gemcitabine will likely become the new standard of care chemotherapy and will shortly be implemented in national and international PDA treatment guidelines. Apart from the striking antitumour effect of nab-paclitaxel alone in KPC tumours, our group previously discovered a potential synergistic drug–drug interaction of gemcitabine and nab-paclitaxel due to ROS-mediated degradation of cytidine deaminase that resulted in higher levels of activated intratumoural gemcitabine metabolites.19 However, the limited availability of patient tissue and the lack of murine albumin-bound paclitaxel have hampered our ability to address the mechanism of action of nab-paclitaxel in human and murine PDA. Importantly, preliminary data suggest that SPARC may play a pivotal role as a biomarker for nab-paclitaxel-based chemotherapeutic regimens and may predict responses to treatment.17
Our results suggest that intratumoural concentrations of paclitaxel following either nab-paclitaxel or cremophor-paclitaxel are comparable in vivo; however, the dramatically reduced toxicity of water-soluble nab-paclitaxel increases the maximum tolerated paclitaxel dose more than fourfold, thus resulting in significantly higher neoplastic cell death rates compared with cremophor-paclitaxel.
To determine whether SPARC would sequester nab-paclitaxel to concentrate the drug intratumourally, we employed two approaches. First, we used equimolar doses of nab-paclitaxel and cremophor-paclitaxel in the KPC model with endogenous SPARC expression and assessed intratumoural paclitaxel concentrations by LC-MS/MS. Second, we genetically ablated SPARC in the closely related KPfC mouse model and used a novel mouse albumin-bound paclitaxel formulation to account for interspecies differences of human albumin and murine SPARC. Both approaches showed that SPARC plays no role in sequestering nab-paclitaxel intratumourally. Despite reports that SPARC is associated with poor survival in PDA patients14 ,15 and that pancreatic tumours grown orthotopically in SPARC−/− mice are more metastatic and less vascular,29 SPARC deficiency did not change the kinetics of tumour onset, growth rate, angiogenesis and metastasis in the KPfC model in our study. However, extensive pharmacokinetic analysis in SPARC+/+ and SPARC−/− mice revealed a SPARC-specific distribution pattern of low-dose m-nab-paclitaxel that was independent from paclitaxel metabolism and saturable at higher concentrations of nab-paclitaxel in tumour-bearing mice. Notably, increased paclitaxel concentrations were observed in plasma and point towards a potential interaction between circulating SPARC and albumin-bound drugs. Alternatively, SPARC has also been shown to be expressed in vascular endothelial cells and platelets,30–32 and has frequently been implicated in the modulation of vascular biology such as endothelial cell proliferation as well as pericyte recruitment and regulation.33–35 It has been previously reported in vitro that endothelial binding and transcytosis of albumin and nab-paclitaxel may be mediated by endothelial cells that express SPARC and other albumin-binding proteins such as gp60.16 ,36 However, using therapeutic concentration of m-nab-paclitaxel (60 mg/kg) in tumour-bearing KPfC mice with genetically ablated SPARC, this effect was saturable and both plasma and intratumoural paclitaxel concentration did not show significant differences in the context of SPARC expression. Also, PDA neoplastic cell apoptosis and tumour volume increases following treatment with m-nab-paclitaxel for 1 week were not substantially affected by SPARC deletion. Therefore, we conclude that although circulating SPARC may directly or indirectly increase the intravascular concentration of low-dose nab-paclitaxel, stromal-derived SPARC does not strongly influence the accumulation of nab-paclitaxel intratumourally in a PDA mouse model. However, we cannot exclude the possibility that extremely high tissue or plasma SPARC concentrations in PDA patients may increase drug concentrations and improve antitumour efficacy, and therefore, the measurement of SPARC plasma levels prior to the initiation of nab-paclitaxel–based chemotherapies should be investigated in pancreatic cancer trials.
Lastly, we examined whether prolonged treatment with m-nab-paclitaxel was able to deplete tumour stroma in KPC mice. Previous work from our group has shown that the extensive desmoplastic reaction of endogenous murine pancreatic tumours can be successfully depleted following 10–14 days of continuous pharmacological inhibition of the sonic hedgehog pathway by IPI-926.10 Therefore, and due to increasing toxicity beyond 2 weeks, we chose to treat tumour-bearing KPC mice over 2 weeks with m-nab-paclitaxel. In contrast to previous data published in patient-derived xenografts,17 we found that m-nab-paclitaxel predominantly induced apoptotic cell death in tumour rather than stromal cells, and the stromal content of desmoplastic murine pancreatic tumours was unchanged following 2 weeks of treatment. This finding should prompt the design of clinical trials that combine nab-paclitaxel with other stromal altering agents such as PEGPH20 (hyaluronidase) to increase intratumoural delivery of nab-paclitaxel.5–12
In conclusion, we present a pharmacokinetic and pharmacodynamic analysis of cremophor-paclitaxel, nab-paclitaxel and a novel m-nab-paclitaxel in various GEMMs of PDA that has potential value for future preclinical and clinical drug development. We found that nab-paclitaxel accumulation and antitumour effects were dose-dependent, but SPARC independent in GEMMs of PDA. Although stromal-derived SPARC may not increase drug accumulation or induce stromal depletion upon nab-paclitaxel treatment in our model system, plasma SPARC levels may play a role in drug retention and therefore tissue delivery in patients with baseline elevated SPARC levels. Future clinical studies will be required to evaluate the potential value of plasma SPARC as a non-invasive and predictive biomarker for nab-paclitaxel-based chemotherapeutic regimens.
Acknowledgments
We thank Manuel Hidalgo for sharing his data before publication. We thank Dr. Aarthi Gopinathan for reviewing ultrasound scans. We thank Frances Connor, Paul Mackin, Lisa Young and Steven Kupczak for maintenance and management of mouse colonies, as well as staff from the Cambridge Research Institute Biological Resource Unit, histology core and pharmacokinetics core.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
Footnotes
-
Contributors AN, KKF and DAT conceived and designed the experiments. AN, KKF and DSC performed animal experiments. AN and KKF performed cell culture experiments. AN, DSC, WJH and VE performed histology stainings. TEB, FMR and DIJ designed and carried out paclitaxel pharmacokinetic experiments. AN and DAT wrote the manuscript. All authors reviewed the manuscript.
-
Funding This research was supported by the University of Cambridge and Cancer Research UK, The Li Ka Shing Foundation and Hutchison Whampoa Limited and the National Institute for Health Research Cambridge Biomedical Research Centre. KKF and DAT were supported by the European Community Grant EPC-TM-Net 256974. AN was supported by the Deutsche Krebshilfe Mildred Scheel Postdoctoral Fellowship. DIJ is a Group Leader in the Cancer Research UK Cambridge Institute. TEB and FMR are supported by Cancer Research UK. DAT is also supported by the Lustgarten Foundation for Pancreatic Cancer Research and by the Cold Spring Harbor Laboratory Association.
-
Competing interests nab-Paclitaxel and m-nab-paclitaxel were formulated and provided by Celgene Corporation.
-
Provenance and peer review Not commissioned; externally peer reviewed.