Article Text

Abstract
Background: Patients with Crohn’s disease have defects in intestinal epithelial permeability that are inadequately explained by known inflammatory bowel disease (IBD) susceptibility genes. E-cadherin (CDH1) plays a vital role in maintaining the integrity of the intestinal barrier and its cellular localisation is disrupted in patients with Crohn’s disease.
Aim: To determine if polymorphisms in the CDH1 gene are associated with Crohn’s disease and to determine the function associated with these polymorphisms.
Methods: The hypothesis was tested using a candidate gene approach using 20 Tag SNPs derived from the HapMap and Crohn’s disease trios. Functional studies were carried out using HapMap cell lines and polarised epithelial cell lines (MDCK-1 and Caco2).
Results: Here we show that CDH1 is associated with Crohn’s disease in 327 trios (rs10431923 excess transmission of “TT” genotype; p = 0.0020) and is replicated in the Wellcome Trust Case Control Consortium CD data set (TT risk allele; OR 1.2, p = 0.005). Patients with the Crohn’s disease risk haplotype (rs12597188, rs10431923 and rs9935563; GTC allelic frequency 21%; p = 0.000016) exhibited increased E-cadherin cytoplasmic accumulation in their intestinal epithelium which may be explained by the presence of a novel truncated form of E-cadherin. Accordingly, expression of this truncated E-cadherin in cultured polarised epithelial cells resulted in abnormal intracellular accumulation and impaired plasma membrane localisation of both E-cadherin and β-catenin.
Conclusion: The mis-localisation of E-cadherin and β-catenin may explain the increased permeability seen in some patients with Crohn’s disease. Thus, the polymorphisms identified in CDH1 are important for understanding the pathogenesis of Crohn’s disease and point to a defect in barrier defence.
Statistics from Altmetric.com
The cadherin super-family of proteins plays an important role in the maintenance of cell polarity and differentiation. E-cadherin is the principal mediator of cell adhesion between intestinal epithelial cells and is found at the adherens junctions (AJs). The AJs and the tight junctions form the apical junctional complex (AJC) that is responsible for the normal barrier function in the intestine.1 Underlying defects in the AJC have long been hypothesised to cause the increased mucosal permeability observed in patients with inflammatory bowel disease.2 3 Recently, we found that PTPsigma, a tyrosine phosphatase that regulates E-cadherin in the colon, is associated with ulcerative colitis,4 and others have shown the AJC protein MAGI2 is associated with inflammatory bowel disease (IBD),5 6 highlighting the importance of the AJC in the pathogenesis of IBD.
There is substantial evidence from both animal models and human observational studies that E-cadherin plays a role in the pathogenesis of IBD. Using a dominant-negative N-cadherin mutant to disrupt E-cadherin function in mice, Hermiston and Gordon demonstrated that loss of E-cadherin produced transmural intestinal inflammation similar to Crohn’s disease.2 These chimeric mice developed colitis with an intact immune system,2 through disruption of the cell–cell and cell–matrix contacts.7 In patients with IBD, E-cadherin is expressed in the intercellular junctions with increased detection in the cytoplasm especially in the areas of regenerating epithelia.8–10 It is also upregulated in areas of active inflammation.11 There is also evidence that pathogens thought to be involved in the development of colitis downregulate E-cadherin. The intestinal pathogens Bacteroides fragilis12 and Candida albicans13 disrupt the adherens junctions through the cleavage of E-cadherin. Similarly, the newly described adherent invasive Escherichia coli found in patients with Crohn’s disease appears to act through displacement of E-cadherin.14 Therefore, subtle defects in E-cadherin function or expression may contribute to increased pathogenicity of this invasive E coli (and other pathogens) found in patients with Crohn’s disease.
The E-cadherin gene (CDH1) is located on chromosome 16, in the region flanking the inflammatory bowel disease-1 (IBD1) locus. E-cadherin (CDH1), along with IL-4R and CD11B, was considered a likely Crohn’s disease candidate gene on chromosome 16 until NOD2 was confirmed to be associated with susceptibility to Crohn’s disease in IBD1.15 16 However, NOD2 polymorphisms do not fully explain the magnitude of genetic linkage on chromosome 16, and at least two other genes in this region are believed to be associated with Crohn’s disease.17 18 Other original chromosome 16 candidate genes, IL-4R and CD11B, were not found to be associated with Crohn’s disease;19 therefore, further susceptibility genes on chromosome 16 are expected.
In this study we utilised HapMap tag SNPs to determine if CDH1 (E-cadherin) is associated with IBD. We show here that E-cadherin is associated with Crohn’s disease, and demonstrate that patients with the disease-associated SNPs have increased E-cadherin cytoplasmic accumulation likely as a result of a processing defect.
METHODS
Subjects
The discovery cohort consisted of 327 Crohn’s disease nuclear families, with 1015 subjects in all (327 probands with both parents, plus 34 affected siblings). The rs12597188, rs10431923 and rs9935563 single nucleotide polymorphisms (SNPs) were genotyped in 401 subjects (129 probands with ulcerative colitis and both parents plus 14 affected siblings). Subjects were recruited from either the Hospital for Sick Children or Mount Sinai Hospital, Toronto, Canada. All probands had a confirmed diagnosis of Crohn’s disease and fulfilled standard diagnostic criteria.20 Phenotypic characterisation was based on the Montreal classification.21 Definitions of L1 and L3 included disease within the small bowel proximal to the terminal ileum and distal to the ligament of Treitz. Perianal disease included only those patients with perianal abscess and/or fistulae. A subject was considered to be Jewish when at least two grandparents were known to be Jewish.
SNP analysis
International HapMap project22 (http://www.hapmap.org; accessed 20 May 2009) Caucasian (CEU) data were used to select the tag SNPs (MAF >1%) that span the CDH1 gene and flanking regions through the “Tagger” software program.23 Twenty tagged SNPs covering CDH1 (chromosome 16, 67303332 to 674436798) region were captured with r2>0.8. Genotype analysis of samples was done using the Illumina genotyping system at The Centre for Applied Genomics, Hospital for Sick Children, Toronto.
Genetic analysis
Linkage disequilibrium between markers and haplotype structures were first calculated using FBAT (http://www.biostat.harvard.edu/∼fbat/fbat.htm; accessed 20 May 2009)24 25 with haplotypes estimated from unphased input using an accelerated EM algorithm. Haploview (http://www.broad.mit.edu/mpg/haploview/; accessed 20 May 2009)26 was then used to map the linkage disequilibrium patterns across the markers and to determine the distinct haplotype blocks. The genotype data were first analysed to determine the presence of any Mendelian inconsistencies and departures from Hardy–Weinberg equilibrium. Families with more than two Mendelian errors were removed from the dataset. The PBAT genetic analysis package (PBAT Version 3.61, http://www.biostat.harvard.edu/∼clange/downloading/PBAT.htm; accessed 20 May 2009) was used for tests of association under additive, recessive and dominant genetic models. Specifically the FBAT–GEE model, implementing the empirical variance–covariance estimator option that adjusts for the correlation among sibling marker genotypes, was applied to examine for single SNP associations. Haplotype association testing was performed using the HBAT option within the package. Jewish ancestry was adjusted for by treating it as a co-variate within the association analysis models. Phenotypic subgroup association analyses, according to the proband’s disease location were performed for those markers identified as being significant in the initial unstratified transmission analysis. Sub-group analyses included both transmission tests within families utilising PBAT, as well as logistic regression models within the probands. Throughout the report, all p values reported are uncorrected except where stated.
Replication
The replication cohort consisted of 1681 subjects with Crohn’s disease and 10 172 healthy population controls recruited by the Wellcome Trust Case Control consortium27 (http://www.ebi.ac.uk/ega/page.php?page = studies&name = WTCCC; accessed 20 May 2009). All subjects within this cohort were of Caucasian descent and resided within Great Britain at the time of enrolment. Affected subjects had a confirmed diagnosis of Crohn’s disease based on standard criteria with a median age at diagnosis of 26.1 years. Subjects were all genotyped using the Affymetric 500K GeneChip (High Wycombe, UK). A logistic regression model was utilised to examine for association between genotype and disease.27
Immunohistochemistry
Formalin-fixed, paraffin-embedded colonic biopsy samples from genotyped individual with Crohn’s disease were obtained. Location of biopsy samples and disease activity were matched between the two groups by the same blinded pathologist. Streptavidin–biotin indirect immunostaining was used as previously described11 using antibodies against E-cadherin (BD Biosciences, San Jose, California, USA).
Splicing
RNA was isolated from immortal lymphobast cell lines derived from the HapMap NA10861, NA12239, NA06985 and NA10846, NA11993, NA12802 individuals using Trizol (Invitrogen, Carlsbad, California, USA), and cDNA was synthesised using SuperScriptTM III (Invitrogen). Primers were designed using sequences from exon 2 and 10 to examine the role of alternate protein production in the precursor region near (primer sequence is available upon request). Sequences of interest were amplified using Platinum Taq DNA polymerase (Invitrogen). The amplified region was cloned into pGEM vector. DNA was purified and sequenced.
Generation of plasmids
Wild-type (WT) E-cadherin tagged with mCherry at the -COOH terminus (WT E-cadherin–mCherry) was constructed by polymerase chain reaction (PCR) using full-length human E-cadherin as template. The following primers containing HindIII restriction sites were used: 5′ primer GCCAAGCTTATGGGCCCTTGAAGCCGCAGC and 3′primer GCCAAGCTTGTCGTCCTCGCCGCCTCCGTA. The Δ72 was created by using the 5′ primer GCCAAGCTTATGACCCGATTCAAAGTGGGC and resulted in the deletion of the first 72 amino acids. The sequences were amplified using Taq polymerase (Invitrogen), subcloned into pGEM-T vector (Promega, Madison, Wisconsin, USA). The mCherry was inserted into pcDNA3 (Invitrogen) using 5′ HindIII and 3′ EcoRV sites and the final WT and Δ72 E-cadherin expression vectors were generated using the HindIII site. The constructs were verified by DNA sequencing.
Cell culture and transfections
Madin–Darby canine kidney (MDCK-1) cells were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin. Transfections were carried out using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol.
Immunofluorescence confocal microscopy
MDCK cell monolayers were grown on glass coverslips. The cells were fixed 48 h post-transfection, stained and analysed using a LSM 510 Zeiss confocal microscope (Zeiss, Toronto, Canada) as described previously.28 Rabbit anti-calnexin (1:250; Sigma, St Louis, Missouri, USA) and rat anti-ZO-1 (1:750; Chemicon, Billerica, Massachusetts, USA) were used to visualise the endoplasmic reticulum (ER) and tight junctions, respectively. β-Catenin was recognised with anti-β-catenin antibodies (1:750; BD XXXXX) and E-cadherin–mCherry or its truncated mutant were recognised by the mCherry fluorescence.
RESULTS
Association of CDH1 with susceptibility to Crohn’s disease
We successfully genotyped 20 tag SNPs (see supplementary table S1) that gave complete genetic coverage of the E-cadherin gene (CDH1) in 327 Crohn’s disease probands and their families (total 1015 persons; table 1). Family-based association analysis of the trio data set revealed two SNPs: rs10431923 (excess transmission of “TT” genotype; p = 0.0020) and rs12597188 (excess transmission of “GG” genotype; p = 0.037), within the gene that were associated with Crohn’s disease. This relationship was not affected by Jewish ancestry and was not seen in ulcerative colitis (rs10431923 p = 0.06500 and rs12597188 p = 0.7254).
The association between rs10431923 and Crohn’s disease remained significant after Bonferroni correction (20 tests; corrected p = 0.04) although the association with rs12597188 did not. In order to replicate these findings, we used the Wellcome Trust Case Control Consortium (WTCCC) Crohn’s disease database.27 The rs10431923 risk associated genotype “TT” was present in 425/1681 (25.3%) Crohn’s disease subjects compared with 2254/10 172 (22.1%) controls (odds ration (OR) 1.2, p = 0.005). The SNP rs12597188 was not included within the Wellcome Trust dataset. Further analysis showed two additional SNPs upstream of CDH1 (rs7186693 excess “G” transmission, p = 0.02; rs1777241 excess “A” transmission p = 0.05) and two additional WTCCC SNPs (not genotyped here) located in the same intron (intron 2) as rs10431923 (rs1078621, p = 0.0075 and rs7203337, p = 0.018) with association to Crohn’s disease, indicating that this region is important in Crohn’s disease.
A number of haplotypes also showed significant association with Crohn’s disease (supplementary table 2, and data not shown) including a 3-SNP haplotype (comprising rs12597188, rs10431923 and rs9935563; GTC allele frequency 21%) that was significantly associated with Crohn’s disease (p = 0.000016; significant after Bonferonni correction for 20 SNPs with three variations – 1140 tests; corrected p = 0.018). Exploration of the disease location sub-groups revealed the association with the rs10431923 T allele was greatest amongst families where the proband had L1 disease (FBAT transmission test: p = 0.009).
Disease-associated SNPs in CDH1 lead to truncations in the processing region of E-cadherin
The identified rs10431923 excess transmitted “T” allele is in strong linkage disequilibrium with over 140 SNPs in the promoter region and flanking precursor region of E-cadherin. Therefore, in order to determine if the risk polymorphism we indentified was a surrogate maker for altered mRNA production, we compared mRNA from genotyped HapMap cell lines homozygous for either the CDH1 GTC risk haplotype (rs12597188, rs10431923 and rs9935563) with the alternate TCA haplotype. The TCA haplotype cell lines had the expected mRNA corresponding to wild-type E-cadherin (fig 1A). However, cell lines with the CDH1 GTC risk haplotype not only had the expected mRNA but also a rare unexpected smaller mRNA corresponding to a novel message that begins at nucleotide 217 of the mature transcript (fig 1A). As seen in fig 1B, the altered mRNA would result in a novel protein encoding a 72 amino acid deletion in the precursor region (N-terminus) of E-cadherin.

Disease-associated single nucleotide polymorphisms (SNPs) results in abnormal E-cadherin production. (A) Polymerase chain reaction (PCR) analysis showing alternate E-cadherin transcripts. Three individuals from the HapMap (CEU NA10861, NA12239 and NA06985) with homozygote disease associated haplotype (GTC) SNPs (rs12597188, rs10431923 and rs9935563) were compared with three individuals (CEU NA10846, NA11993, NA12802) with homozygote nucleotide non-disease associated haplotype TCA SNPs (rs12597188, rs10431923 and rs9935563). After digestion with EcoR1, the PCR product isolated from the control individuals (right) had two equivalent size fragments of approximately 1.2 kb corresponding to wild-type E-cadherin, while individuals with the disease-associated haplotype (left) had two fragments of 1.2 and 1.1 kb corresponding to the alternate truncated E-cadherin. (B) Schematic representation of the novel alternate (N-terminally truncated) E-cadherin. Individuals with the Crohn’s disease risk haplotype have an abnormal truncated protein: the normal (wild-type protein, top) and the abnormal truncated protein missing S and part of Pre domains (bottom). Cad, cadherin domain; ICD, intracellular domain; Pre, precursor domain (both S and Pre are normally cleaved after E-cadherin is transported to the plasma membrane); S, signal peptide region; TM, transmembrane domain.
Disease-associated SNPs lead to cytoplasmic accumulation of E-cadherin in the intestinal epithelium of patients and in transfected epithelial cells
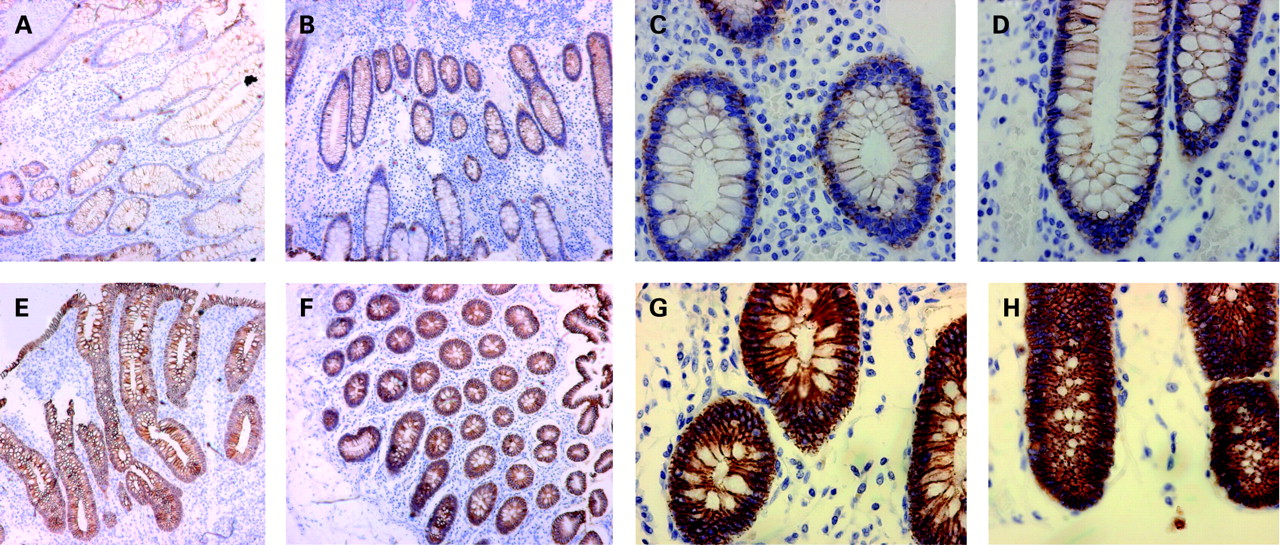
To determine if the GTC risk haplotype contributed to abnormal expression or cellular localisation of the E-cadherin protein, we compared E-cadherin expression in biopsies from four paediatric patients homozygous for the GTC risk haplotype with four paediatric patients homozygous for the CAG genotype. These patients were matched for disease location and degree of disease activity by the pathologist (BN). As seen in fig 2, individuals with the GTC risk haplotype had increased E-cadherin staining throughout the epithelia, and in particular, they exhibit strong intracellular accumulation of E-cadherin when compared with the TCA genotype.

E-cadherin staining in the intestine of patients with Crohn’s disease. (A–D) Intestinal biopsies from patients without Crohn’s disease associated CDH1 SNP haplotype. E-cadherin (brown staining) was found to be localised to the plasma membrane of enterocytes. (E–H) Intestinal biopsies from patients with Crohn’s disease disease associated CDH1 SNP haplotype. E-cadherin (brown staining) exhibits increased cytoplasmic accumulation in the enterocytes. (A, B, E, F original: ×10 magnification; C, D, G, H original: ×40 magnification).
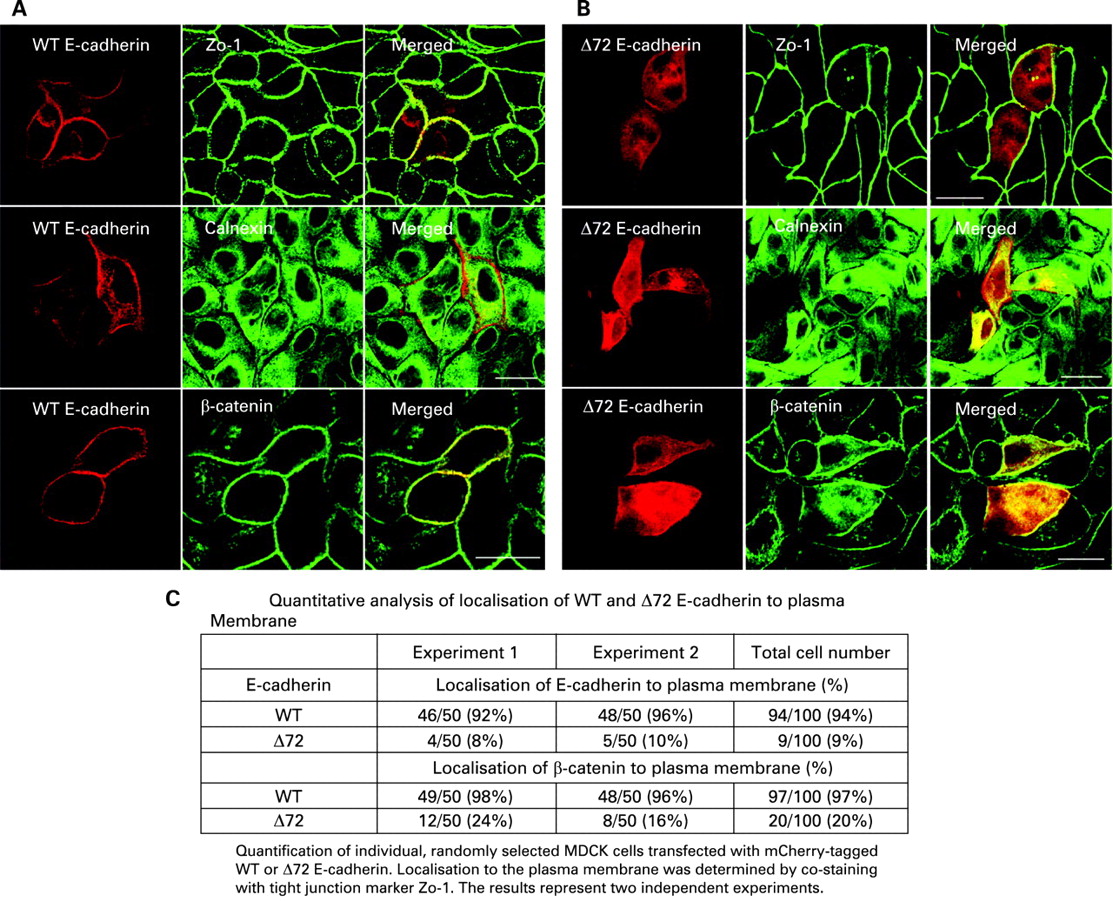
The precursor domain of E-cadherin is proposed to prevent aggregation of the protein prior to trafficking to the plasma membrane.29 To determine if the alternate protein identified resulted in cytoplasmic aggregation and retention similar to the E-cadherin staining observed in patients with the disease associated SNPs, we transfected wild-type and the N-terminal truncated E-cadherin, tagged with mCherry, into polarised epithelial MDCK cells. As shown in fig 3A (and quantified in fig 3C), the wild-type E-cadherin was localised to the plasma membrane, as demonstrated by co-localisation with the junctional protein marker ZO-1. In contrast, the truncated protein accumulated in the cytoplasm (fig 3B,C), as determined by co-localisation with the endoplasmic reticulum marker protein calnexin. Similarly, endogenous β-catenin co-localised with wild-type E-cadherin at the plasma membrane, as expected, but in cells transfected with the truncated form of E-cadherin, β-catenin was mis-localised and accumulated in the cytoplasm, much like the truncated E-cadherin (fig 3B). Identical results were also obtained using mouse wild-type and truncated E-cadherin (supplementary fig s1) and in human intestinal polarised epithelial cell line Caco2 (supplementary fig s2).
{kind=link}
{kind=link}
{kind=link}
Cytoplasmic accumulation of Δ72 E-cadherin in transfected MDCK cells. (A,B) Immunofluorescence (confocal) analysis was carried out to determine the cellular localisation of E-cadherin and the Δ72 mutant. MDCK cells expressing mCherry-tagged (red) (A) WT E-cadherin, and (B) Δ72 E-cadherin co-stained with tight junction marker ZO-1 (green), ER marker Calnexin (green) or β-catenin (green). The upper panels of (A) and (B) represent the apical view to visualise apical junctional complexes, while the middle and lower panels represent the middle of the cell. (A and B) are representative single plain images of 50 cells scored in each of two separate experiments (table, inset). Scale bars represent 20 μm. (C) Quantitative analysis of localisation of WT and Δ72 E-cadherin to the Plasma Membrane. ER, endoplasmic reticulum; WT, wild-type.
Interestingly, in both intestinal epithelia from Crohn’s disease patients with the risk associated polymorphism and in cell culture transfected with the corresponding truncated form of E-cadherin, the architecture of the epithelium became disorganised (fig 2E,F and fig 3, and supplementary figs s1 and s2). Collectively, these data demonstrate that Crohn’s disease-associated SNPs in E-cadherin lead to N-terminal truncations of the protein, resulting in its cytoplasmic accumulation and inability to reach the plasma membrane.
DISCUSSION
Although NOD2 is the major susceptibility gene for Crohn’s disease located at IBD1, a recent genome-wide scan18 has confirmed that at least two other genes in this region are likely to be associated with Crohn’s disease.17 Here we show that CDH1 (E-cadherin), which is located within the IBD1 locus, is associated with Crohn’s disease. We found that homozygous carriage of the CDH1 rs10431923 T allele was associated with Crohn’s disease.
Although we found a nominal association in the Wellcome Trust Case Control Consortium database,27 this association was not published in any of the recent genome-wide association studies.27 30–32 This is not unexpected as the genes found in those studies only account for 20% of the hereditary risk of Crohn’s disease33 and the stringent statistical corrections applied may have resulted in a type II error with true associations being missed.34 Certainly, a number of candidate gene studies with strong functional data have shown association with genes not identified in these genome-wide association (GWA) studies.4 5 35 Furthermore, the genetic association for E-cadherin is based on recessive modelling that is not well captured by GWA studies, which utilised a case–control design.
The SNPs identified here are found in introns. A number of intronic polymorphisms in these regions of E-cadherin are known to cause similar deletions in the precursor domain as described here.36 Furthermore, intron 2 is known to have a number of cis-regulatory elements that are critical for CDH1 gene regulation.37 The novel isolated mRNA is very similar to previously described expressed sequence tag (EST) transcripts AK311198, AK309703 and DC370126. This altered mRNA may be due to alternate splicing or alternate translation start sites associated with these polymorphisms. Alternate translation in N-terminal is known to result in the synthesis of N-terminal truncated proteins that may have important cellular function including altered subcellular targeting,38 39 very similar to what we observed here. In support of this proposed processing defect, patients with Crohn’s disease with the disease associated SNPs showed increased cytoplasmic E-cadherin staining. The precursor domain is thought to prevent E-cadherin from aggregating in the cytoplasm prior to trafficking to the cell membrane, and accurate processing of this precursor domain is required for E-cadherin adherent functions at the adherens junction.29 Accordingly, we show an accumulation of the truncated protein in the cytoplasm instead of the expected plasma membrane localisation in polarised cultured epithelial cell lines.
It is interesting to speculate how these identified polymorphisms in the E-cadherin gene may influence the onset of Crohn’s disease. In experimental E-cadherin knockout mice, complete loss of E-cadherin was shown to be lethal; however, loss of 50% of E-cadherin (heterozygous mice) resulted in normal mice with normal adherens junctions.40 Therefore, the truncated E-cadherin described here would not be expected to be the predominant form and may only be expressed in certain unknown conditions. However, this truncated E-cadherin may interfere with the normal E-cadherin function by aggregating and preventing some of the protein from reaching the plasma membrane, resulting in morphological abnormalities of the intestinal epithelium that we observe in the human tissue samples and in transfected epithelial cells. This may also destabilise the adherens junction and allow bacteria or bacterial products to enter the intestinal submucosa, and this, coupled with polymorphisms in innate immunity such as NOD2, ATG16L1 or IRGM, could lead to the onset of Crohn’s disease. This effect may be more pronounced during times when E-cadherin production is expected to be upregulated, such as during infection or inflammation. Similarly, E-cadherin is known to be downregulated by pathogenic bacteria, such as the Crohn’s disease invasive E coli.14 Therefore, a weakened adherens junction may allow for increased pathogenicity of these (and other) Crohn’s disease associated bacteria. Interestingly, the truncated E-cadherin not only disrupted E-cadherin trafficking to the plasma membrane, but also β-catenin subcellular localisation. β-Catenin, which normally associates with E-cadherin and helps connect it to the actin cytoskeleton, is known to be involved in regulation of the crypt–villous axis of epithelial cell lineage differentiation and in intestinal adenoma formation;41 therefore, mis-localisation of β-catenin may result in abnormalities in the crypt–villous axis that predisposes patients to colitis. The cytoplasmic accumulation of E-cadherin observed here is very similar to the cytoplasmic staining of the misfolded MUC2 mucin protein reported in both humans and a mouse model of colitis.42 The mucin study, along with a recent XBP1 knockout mice model of colitis and a human IBD genetic study, show that endoplasmic reticulum (ER) stress may be important in the development of IBD.35 42 Although we did not find altered expression of grp78 and XBP1 (data not shown), it is possible that the abnormal cytoplasmic accumulation of E-cadherin results in a similar ER stress that may, along with disruption of the AJs, be involved in the pathogenesis of IBD in patients with disease-associated polymorphisms in CDH1.
Intestinal barrier defects causing increased permeability have been proposed as a primary contributor in the pathogenesis of IBD.43–45 This is supported by studies that show permeability defects in patients with IBD,46–49 prior to relapse,48 and even prior to diagnosis.50 The adherens junctions and the tight junctions form the apical junctional complex (AJC) that is responsible for normal intestinal barrier function.1 Underlying defects in the AJC have been hypothesised to cause the increased mucosal permeability observed in patients with IBD.2 3 Recently, polymorphisms within AJC genes have been shown to play a pivotal role in the pathogenesis of IBD through the disruption barrier defence,4 6 51 52 and point to an important role for E-cadherin. We have previously shown that a polymorphism within the PTPRS gene (a tyrosine phosphatase that dephosphorylates E-cadherin) results in alternate splicing that removes an immunoglobulin domain of the PTPsigma protein and is associated with an increased susceptibility to ulcerative colitis.4 Similarly, MAGI2, which encodes membrane-associated guanylate kinase inverted-2 protein that is thought to act as a scaffolding protein and interacts with β-catenin (which binds directly to E-cadherin at the adherens junctions53 54), has been shown to be associated with ulcerative colitis and coeliac disease.6 Finally, the DLG5 gene,55 found to be associated with both ulcerative colitis and Crohn’s disease,54 is also involved in regulation of adherens junctions. Notably, discs large (Drosophila homolog) (DLG)5 has been recently shown to target and stabilise the β-catenin:E-cadherin complex at AJ.56
In summary, our results demonstrate an association between polymorphisms within the CDH1 (E-cadherin) gene and Crohn’s disease, which leads to cytoplasmic accumulation of E-cadherin instead of its normal localisation at the plasma membrane/adherens junction. These data further underscore the importance of both E-cadherin and the integrity of the intestinal epithelial junctions in the development of Crohn’s disease. Further studies of E-cadherin and its association with other known IBD genes will advance understanding of the pathogenesis of Crohn’s disease.
Acknowledgments
We thank J Stempak, C Lu, K Lau, C H Guo and Ch Bruce for technical support. Thanks to Drs D Turner and A-C Villani for critical reading of this manuscript.
REFERENCES
Supplementary materials
Web only appendix 58;8:1121
Files in this Data Supplement:
Footnotes
Competing interests: None.
Funding: This work was supported by the Canadian Institute of Health Research (CIHR, grant no MOP-86496) to DR, NIDDK Grant (DK-06-504) to MS, the Crohn’s Colitis Foundation of Canada (CCFC) to AMG and DR, and a Thrasher Research Fund New Investigators Grant to AMM. AMM is supported by a transition award from the CCFC/CAG/CIHR and a Canadian Child Health Clinician Scientist Program (Strategic Training Initiatives in Health Research Program – CIHR) award. TW is supported by CCFC and AstraZenca Partnered fellowships from the CAG/CIHR. MSS is supported by the Gale and Graham Wright Research Chair in Digestive Diseases at Mount Sinai Hospital. DR holds a Canada Research Chair (Tier I) from the CFI/CIHR.
Ethics approval: Study subject phenotypic information and DNA samples were obtained with institutional review board approval for IBD genetic studies at the Hospital for Sick Children and Mount Sinai Hospital in Toronto. Written informed consent was obtained from all participants.
▸ Two supplementary figures and two supplementary tables are published online only at http://gut.bmj.com/content/vol58/issue8