Article Text

Abstract
Background and aims: Administration of interferon (IFN)-β may represent a rational approach to the treatment of ulcerative colitis through its immunomodulatory and anti-inflammatory effects. The present study was performed to evaluate the efficacy and tolerability of IFN-β-1a.
Methods: Patients (n=18) with moderately active ulcerative colitis were randomised to receive IFN-β-1a or placebo. IFN-β-1a was started at a dose of 22 μg three times a week subcutaneously, and the dose was increased at two week intervals to 44 μg and then to 88 μg if no response was observed. The maximum duration of treatment was eight weeks. End points were clinical treatment response, defined as a decrease of at least 3 points from baseline in the ulcerative colitis scoring system (UCSS) symptoms score and induction of endoscopically confirmed remission.
Results: Baseline characteristics and disease severity were similar in both groups. Data from 17 patients are included in this report (10 patients in the IFN-β-1a group and seven patients in the placebo group). Clinical response was achieved in five patients (50%) in the IFN-β-1a group and in one (14%) in the placebo group (P=0.14). Remission was achieved in three patients in the IFN-β-1a group and in none in the placebo group (p=0.02). Most adverse reactions associated with IFN-β-1a were influenza-like symptoms or injection site reactions, and were mild or moderate in severity.
Conclusions: IFN-β-1a may represent a promising novel treatment approach in ulcerative colitis.
- cytokines
- inflammatory bowel disease
- ulcerative colitis
- interferon
- IFN, interferon
- IL, interleukin
- TNF, tumour necrosis factor
- UCSS, ulcerative colitis scoring system
- PGA, physician’s global assessment
Statistics from Altmetric.com
- IFN, interferon
- IL, interleukin
- TNF, tumour necrosis factor
- UCSS, ulcerative colitis scoring system
- PGA, physician’s global assessment
Although the aetiology of ulcerative colitis has not been fully elucidated, immunological factors are believed to play an important role. Plasma and tissue concentrations of proinflammatory cytokines, including interferon (IFN)-γ, interleukins (IL)-1β, 6 and 8, and tumour necrosis factor (TNF), are increased in patients with ulcerative colitis.1–5 Levels of these cytokines have been shown to be related to disease activity.6 Such findings suggest that immunomodulatory therapy might be beneficial in the treatment of ulcerative colitis. However, although there is evidence that immunosuppressive therapy with agents such as azathioprine or 6-mercaptopurine may prolong remission,7–10 anti-inflammatory treatment with aminosalicylates or corticosteroids remains the mainstay of ulcerative colitis management.11
Immunomodulatory therapy with IFN-β represents a potentially useful new treatment strategy in ulcerative colitis due to the diverse effects of this cytokine on immunological and inflammatory processes. IFN-β has been shown to inhibit the production of IFN-γ and TNF,12,13 and to antagonise early events in the IFN-γ signalling pathway.14 In addition, IFN-β increases expression of the anti-inflammatory cytokine IL-10,15 and enhances T suppressor and natural killer cell activity.16,17 Antiviral effects of interferons result from induction of the enzyme 2′-5′oligoadenylate synthetase as well as protein kinase C. These two enzymes also convey antiproliferative and cell growth inhibitory activities.18 Other important effects of interferons include a protective action against bacterial and parasitic infections, which has been demonstrated in various model systems.19–22
An open study with another type I interferon, IFN alpha, obtained a remission rate of 82% in patients with refractory ulcerative colitis after six months of treatment (3–9 million units/thrice weekly subcutaneously),23 but few studies to date have examined the effect of IFN-β. One exception is a recent open pilot study in which remission was achieved in 22 of 25 patients treated three times a week with IFN-β 0.5 or 1 million U, for a mean of one year.24 The present study was performed to investigate the efficacy and tolerability of subcutaneous administration of recombinant IFN-β-1a in the treatment of moderately active ulcerative colitis.
MATERIALS AND METHODS
The trial was a randomised, double blind, intraindividual, dose escalating study performed at six centres in Belgium, Canada, and Germany. It was conducted from October 1998 to March 2000 according to the principles of Good Clinical Practice and the Declaration of Helsinki, and was approved by institutional review boards or local ethics committees at each centre. Written informed consent was obtained from all patients prior to entry into the study.
Patients
Patients were eligible for inclusion in the study if they were at least 18 years of age and had moderately active ulcerative colitis, as defined by a score of 6–10 on the ulcerative colitis scoring system (UCSS),25 with a proctosigmoidoscopy score of 2. The UCSS is a combination of rating scales for stool frequency, rectal bleeding, endoscopic activity, and physician’s global assessment (PGA) (each 0–3 for no activity–severe disease). The maximum total UCSS score is 12 for severe disease.25 Patients were also required to have an adequate bone marrow reserve (white cell count ≥3.5×109/l, neutrophils >1.5×109/l, thrombocytes ≥100×109/l and ≤800×109/l, and haemoglobin ≥9 g/dl). Female patients were required to be either postmenopausal or surgically sterile, or to be using adequate contraception.
Exclusion criteria included severe ulcerative colitis, defined as a UCSS PGA score of 3, planned or emergency surgery, previous interferon therapy, or cytokine/anticytokine therapy within the previous six months. Patients were also ineligible if they had inadequate liver or renal function, a history of cancer (other than basal cell carcinoma), active infectious disease, other serious medical conditions, or a history of alcohol or drug abuse.
Concomitant therapies
Only stable oral doses of 5-ASA were allowed (up to 3 g/day, stable for eight weeks prior to inclusion, no topical rectal treatment) as concomitant and prior therapy. The following treatments were not allowed during the study treatment and had to be discontinued before inclusion: immunosupressives (azathioprine/6-mercaptopurine, methotrexate, cyclosporin A, all to be discontinued at least 12 week prior to randomisation), antibiotics (discontinued two weeks prior), antiperistaltic medication (that is, loperamide or opiates, discontinued two weeks prior), and non-steroidal anti-inflammatory drugs except paracetamol (discontinued two weeks prior). Prestudy use of glucocorticoids was permitted only with a maximum of two single doses within the four weeks prior to randomisation. Any investigational drug or colitis relevant experimental procedure within four weeks prior to the study was forbidden.
Protocol and end points
Eligible patients were randomised by means of a computer generated list produced at the Corporate Biometrics Department of Serono International SA (Geneva, Switzerland) to receive either IFN-β-1a (Rebif; Serono) or placebo. Randomisation was stratified by centre with a block size of three (2:1 IFN-β-1a:placebo). Treatment with IFN-β-1a was started at a dose of 22 μg three times a week subcutaneously. Improvement was defined as a decrease of 1 point in the combined score of UCSS symptoms and PGA. If no improvement was observed after six injections, the dose was increased to 44 μg (three times a week subcutaneously), and increased further to 88 μg (three times a week subcutaneously) if no improvement was observed after six injections with the 44 μg dose. If improvement was observed after six injections at any dose, the patient entered a maintenance treatment phase of 6–12 injections at that dose. If no improvement was observed after six injections at 88 μg, or if remission occurred at any point, treatment was stopped. Details of the dose rising scheme are detailed in fig 1. The treatment period was concluded by a four week follow up phase after the last study drug administration.

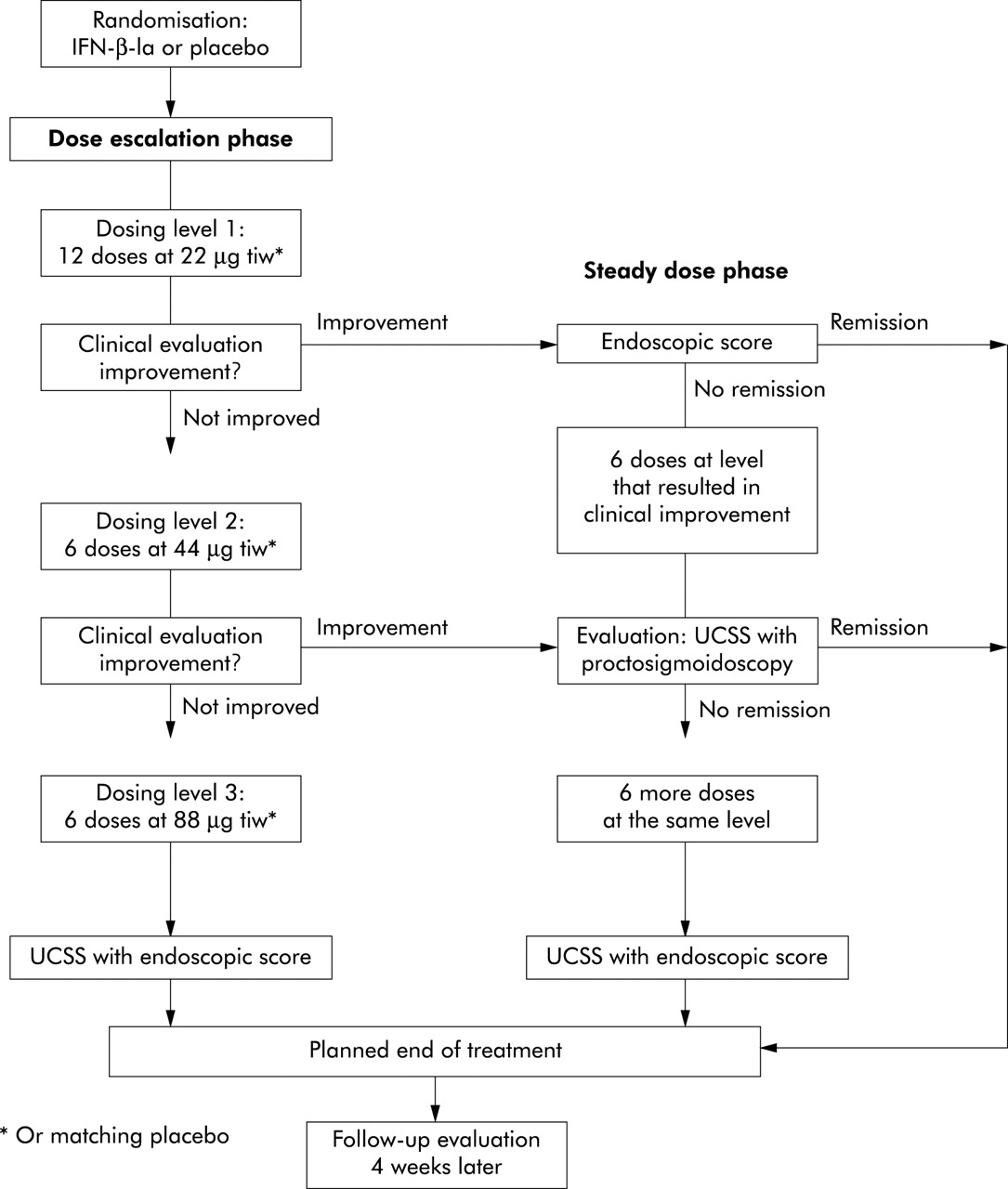{kind=link}
Study flow chart. All patients received 22 μg of study drug three times a week (tiw) for four weeks. If patients did not show clinical improvement the dose was doubled every two weeks to up to 88 μg tiw. “Improvement” was defined as a decrease in the clinical component of the ulcerative colitis scoring system (UCSS) score (that is, stool frequency, rectal bleeding, and physician’s global assessment) by at least 1 point. If clinical symptoms improved, a sigmoidoscopy was performed. If patients were in remission by the combined UCSS score, a study end point was reached. Otherwise, the treatment dose was kept stable for two or four more weeks, respectively. The maximum duration of treatment was eight weeks and the minimum duration was four weeks. End points: remission was defined as a UCSS score of 0 for the clinical component as well as a score of 0 or 1 in the endoscopic part of the UCSS score. Clinical response was defined as a decrease of 3 or more points from baseline in the clinical components of the UCSS score. Patients discontinued due to adverse events were advanced to study end examination, including complete assessment of the UCSS.
Efficacy was assessed by means of the UCSS every two weeks during treatment, at the end of the treatment period, and at the end of the four week follow up period. Efficacy end points were treatment response and remission. Treatment response was defined as a decrease of at least 3 points from baseline in the UCSS symptoms score and PGA (without the proctosigmoidoscopic score) during treatment. This was a modification of the end point in the original protocol, which defined a clinical response as a decrease of at least 1 point in the UCSS symptoms score and PGA. Remission was defined as complete resolution of clinical symptoms (all clinical UCSS subscores equal to 0), with a proctosigmoidoscopy score of 0 or 1 at any time during treatment. Secondary end points included overall treatment and end point responses (defined as a decrease in UCSS symptoms score, PGA, and proctosigmoidoscopic scores of at least 1 point during or at the end of treatment), and clinical end point responses (a decrease of at least 1 point from baseline in UCSS symptoms scores and PGA, without the proctosigmoidoscopic score).
Information on adverse events was collected throughout the study. All events were graded according to the modified World Health Organization (WHO) Recommendations for Grading of Acute and Subacute Toxicities (grade 1, mild; grade 2, moderate; grade 3, severe; grade 4, life threatening). Blood samples for standard haematology and clinical chemistry investigations were obtained at the start of treatment, at the end of each dose escalation period, and at the end of the follow up period. In addition, serum samples for measurement of neutralising antibodies to IFN-β-1a were obtained at the end of the follow up period. Neutralising antibodies in these samples were measured by enzyme linked immunosorbent assay.
The trial design was exploratory. Therefore, no formal sample size calculation was performed. Response/remission rates were compared between the two treatment groups by one sided Fisher’s exact test.26 All analyses were performed on an intention to treat basis, with missing data being replaced according to the last observation carried forward principle.
RESULTS
Study population and patient disposal
Of the 18 patients enrolled, data from 17 patients were analysed. One patient was excluded a priori (that is, before the code was broken) from the analysis because of misallocation of study drug. Seven of 17 patients were randomised to receive placebo and 10 to receive IFN-β-1a. Baseline demographics of the study population were well balanced with regard to sex, age, and disease characteristics (table 1). The median UCSS score was 9 for patients receiving IFN-β-1a and 8.5 for patients receiving placebo. There were no significant differences between treatment groups in the use of concomitant therapies prior to initiation of the study drugs. During the course of the study, six patients (four in the IFN-β-1a group, two in the placebo group) stopped treatment because of progressive disease, and two (both in the IFN-β-1a group) for other reasons. All patients were included in the intention to treat and safety analyses.
Patient demographics and disease characteristics in the two groups
The median duration of treatment was 35.5 days. Of the 10 patients on subcutaneous interferon-β-1a, four were escalated to the maximum dose of 88 μg three times a week, two reached 44 μg, and four remained on 22 μg. The average dose for IFN-β-1a treated patients was 38.6 μg three times a week, which represents an average daily dose of 16.7 μg IFN-β-1a and a cumulative total dose of 627 μg IFN-β-1a.
Efficacy
Clinical response, as defined by a decrease of at least 3 point in the UCSS, was achieved in five patients (50%) in the IFN-β-1a group and in one (15%) in the placebo group (p=0.14). Endoscopically confirmed remission was achieved in three patients in the IFN-β-1a treated group and in none in the placebo group (p=0.02).
Of the five patients in whom clinical responses were achieved during IFN-β-1a treatment, one was receiving 22 μg three times a week, three were receiving 44 μg, and one was receiving 88 μg. The mean time to clinical treatment response was 28 (SD 11) days in the IFN-β-1a group and 28 days in the responding placebo patient. The mean cumulative dose of IFN-β-1a required to achieve clinical treatment response was 449 μg.
Of the three patients in whom remission was achieved during IFN-β-1a treatment, one was receiving 44 μg and two were receiving 88 μg. The mean time to remission was 52 (7) days in the IFN-β-1a group. The mean cumulative dose of IFN-β-1a required for induction of remission was 1115 μg.
UCSS symptom scores and PGAs tended to decrease to a greater extent in patients treated with IFN-β-1a than in those receiving placebo, but the differences were not statistically significant (table 2).
Change in UCSS symptom scores from baseline, and response and remission rates during treatment in the two groups
Safety
A total of 92 adverse events were reported, of which 57 occurred in the IFN-β-1a group and 35 in the placebo group; all patients experienced at least one adverse event. The majority of adverse events (97%) were graded as mild or moderate in severity; only one adverse event in the IFN-β-1a group (pain related to disease progression) was rated as severe.
Adverse events that were considered to be possibly or probably treatment related occurred in 15 patients (10 in the IFN-β-1a group; five in the placebo group). Most were influenza-like symptoms or injection site reactions, and most were mild or moderate in severity (table 3). One patient in the IFN-β-1a group withdrew from the study because of treatment related side effects (influenza-like symptoms).
Adverse events considered possibly related to treatment
Five patients in the IFN-β-1a group showed an increase in lymphocyte counts to at least WHO grade 3 before starting treatment; in three of these, lymphocyte counts had returned to values similar to before the start of study treatment by the end of the study. Three patients (two in the IFN-β-1a group, one in the placebo group) showed a 2 point increase in WHO grade for some blood chemistry variable during the study. These were graded by the investigators as without major clinical impact. No other significant laboratory abnormalities (based on WHO classifications) were observed. All patients were tested for neutralising antibodies before the study and after the end of follow up. No neutralising antibodies to IFN-β-1a were detected.
DISCUSSION
The results of this pilot study suggest that subcutaneous application of IFN-β-1a may be beneficial in the treatment of moderately severe ulcerative colitis. Patients treated with escalating doses of IFN-β-1a tended to show higher clinical response and remission rates than those receiving placebo, although the differences between the groups did not reach statistical significance. It should be noted however that this was an exploratory study that was not powered to detect significant differences in response rates. Allocation of patients showed some imbalances between the two groups (in sex distribution and disease localisation), which are to be expected in a small trial but which may also have influenced the outcome.
The mean dose of IFN-β-1a required to induce a new clinical treatment response was 449 μg subcutaneously, given for approximately 28 days. This is equivalent to a mean dose of 35 μg given three times a week. The mean dose required to induce remission was 1115 μg over a mean of 52 days, which corresponds to a dose of 50 μg given three times a week. These may be considered the minimal effective doses for use in ulcerative colitis, and are comparable with those previously shown to be effective in patients with multiple sclerosis.23–29
Subcutaneous application of IFN-β-1a was well tolerated in this study. The majority of adverse events were mild or moderate, and consisted of influenza-like symptoms and injection site reactions. Thus the adverse event profile of IFN-β-1a in these patients with ulcerative colitis was comparable with that seen previously in large studies in multiple sclerosis patients.27,28
The finding that subcutaneous application of IFN-β-1a can induce clinical responses in patients with ulcerative colitis is consistent with those of open studies with IFN alpha in this condition,23,30 and of a long term pilot study with IFN-β.25 This raises important questions about the potential mechanisms of action of IFN-β in ulcerative colitis. In vitro studies have shown that IFN-β can induce IL-10 release from lymphocytes obtained from patients with multiple sclerosis,15,25 which suggests that IFN-β could induce an anti-inflammatory response in the colonic mucosa. Furthermore, a study in patients with Crohn’s disease and concomitant herpes virus infection has shown that IFN alpha can induce an antiviral reaction that was associated with reduced intestinal inflammation.31 At present however the clinical significance of such findings remains to be established.
In conclusion, mild to moderate uncomplicated ulcerative colitis is a condition that favours exploration of the efficacy of IFN-β-1a in this disease. However, the side effect profile of IFN-β-1a suggests that it might find its place in the therapy of more serious disease as an alternative to glucocorticoids, if effective. Further studies are warranted to confirm and extend these findings, and to define the potential benefits offered by IFN-β as an alternative to glucocorticoids.
Acknowledgments
The clinical study was supported by a grant from Serono International SA, Geneva, Switzerland. The fellowship of SN was supported by the Competence Network “Inflammatory Bowel Disease” (a public program financed by the German Ministry of Education and Research (BMBF)) and a grant from Mucosa Forschungsgesellschaft (MFG).
REFERENCES
Linked Articles
- CORRECTION