Article Text

Abstract
Background and aims: Chromosomal instability is one of the most consistent markers of sporadic colorectal cancer in humans. There is growing evidence that telomere shortening is one of the mechanisms leading to chromosomal instability and cancer initiation.
Methods: To test this hypothesis, the telomere length of colorectal epithelial cells and cells from connective tissue was determined at the adenoma-carcinoma transition at the cellular level by quantitative fluorescence in situ hybridisation.
Results: Our study showed that the telomere fluorescence intensity of epithelial cells was significantly weaker at the earliest morphologically definable stage of carcinoma—high grade dysplasia with minimal invasive growth—compared with the surrounding adenoma. In contrast, cells from connective tissue had a similar telomere signal intensity at the carcinoma stage compared with the adenoma, and in turn cells from connective tissue had overall significantly stronger telomere fluorescence signals compared with epithelial cells.
Conclusions: These results demonstrate that short telomeres of epithelial cells characterise the adenoma-carcinoma transition during human colorectal carcinogenesis, suggesting that carcinomas arise from cells with critical short telomeres within the adenoma. Since the adenoma-carcinoma transition in colorectal cancer is characterised by an increase in chromosomal instability and anaphase bridges, our data support the hypothesis that short telomeres initiate colorectal cancer by induction of chromosomal instability.
- telomere shortening
- adenoma
- high grade dysplasia
- colorectal cancer
- chromosomal instability
- CIN, chromosomal instability
- MIN, microsatellite instability
- Q-FISH, quantitative fluorescence in situ hybridisation
- TFI, telomere fluorescence intensity
Statistics from Altmetric.com
- CIN, chromosomal instability
- MIN, microsatellite instability
- Q-FISH, quantitative fluorescence in situ hybridisation
- TFI, telomere fluorescence intensity
Colorectal cancer is one of the most common malignancies worldwide and is characterised by sequential morphological changes of the colonic mucosa spanning the adenoma-carcinoma transition coupled with increasing genetic alterations.1 Morphologically, the adenoma-carcinoma transition is characterised by the emergence of high grade dysplastic epithelial cells clonally expanding within the surrounding adenomatous polyp. These areas of high grade dysplasia will eventually invade the muscularis mucosa of the intestine and this stage represents the earliest morphologically definable stage of carcinoma during human colorectal carcinogenesis. On a molecular level, the carcinoma stage is characterised by genetic instability which is generally divided into two categories2: (1) chromosomal instability (CIN), characterised by aneuploidy; and (2) microsatellite instability (MIN), characterised by defects in DNA repair. In human colorectal cancer formation, MIN is predominant in familial cases of colorectal cancer formation3,4 (for example, hereditary non-polyposis coli) whereas CIN is prevalent in the vast majority of sporadic cancers in the elderly.2 CIN sharply increases at the adenoma-carcinoma transition.5–14 Mutations in several genes governing mitotic15 and cell cycle checkpoints (for example, p53)16 have been linked to the onset of CIN at the adenoma-carcinoma transition but the mechanisms leading to the CIN phenotype have yet to be evaluated. One current hypothesis is that loss of telomere function triggers CIN thus leading to cancer initiation.17 Experimental merit for this hypothesis has come from studies in the telomerase deficient mouse (mTERC−/−) showing that critical telomere shortening leads to elevated rates of spontaneous cancer during aging18 and increased initiation of aberrant crypt and micro-adenomas during the time course of colorectal cancer formation.19 In addition, telomere shortening in double knockout mice lacking telomerase and p53 provoked an increase in epithelial cancer (breast and colon cancer) characterised by CIN similar to the human epithelial cell cancer.20
Does the telomere hypothesis of cancer initiation apply to human cancer? Support for this hypothesis of cancer initiation has come from studies showing that human cancers contain significantly shorter telomeres compared with the surrounding non-transformed tissue.21–26 Nevertheless, these studies have exclusively analysed DNA from whole tissue samples of advanced cancer stages. Therefore, it remains possible that telomere shortening in cancer tissue is induced by cell proliferation from early stage cancer towards advanced stages, thus simply presenting a coincidence of short telomeres and cancer without a functional link to cancer initiation. In addition, tumours often represent a mixture of cells, including a high percentage of non-transformed supporting cells and endothelial cells, up to 70% in colorectal cancer.25
To directly test the telomere hypothesis of cancer initiation, we have analysed telomere length in a cell type specific manner at the adenoma-carcinoma transition in 10 cases of sporadic colorectal cancer formation by the use of quantitative fluorescence in situ hybridisation (Q-FISH). We show that the telomere fluorescence intensity (TFI) of epithelial cells is significantly weaker at the earliest carcinoma stage—high grade dysplasia with minimal invasive growth—compared with the surrounding adenoma. In contrast, the TFI of connective tissue is overall stronger compared with epithelial cells but with similar signal intensity in adenoma and high grade dysplasia. Together, our data support the hypothesis that telomere shortening of epithelial cells plays a role in the initiation of human colorectal cancer.
MATERIALS AND METHODS
Patient characteristics
Ten samples showing the adenoma-carcinoma transition in cases of sporadic colorectal cancer formation were selected from the pathology archive at the Medical School of Hannover. Patients were aged 65–82 years. All samples showed cases of sporadic polyps of the colorectum diagnosed and excised during routine endoscopy at the endoscopy unit, Medical School Hannover.
Quantitative fluorescence in situ hybridisation (Q-FISH)
Paraffin sections (3 μm) were deparaffinised with xylol and rehydrated. After fixation in 4% formaldehyde, enzymatic unmasking was performed for 10 minutes at 37°C (enzyme mix: 125 mg pepsine/105 μl concentrated HCl/125 ml distilled water). Fixation and washing steps were repeated as described, followed by dehydration of the slides. After three minutes of denaturation at 80°C, hybridisation was carried out for two hours at room temperature in the dark. (Hybridisation mix: 1.5 μl 1 M Tris Cl, pH 7.2, 10.7 μl MgCl2 (25 mM MgCl2/9 mM citric acid/8.2 mM NaH2PO4/pH 7.4), 87.5 μl deionised formamide, 6.2 μl 10% (w/w) blocking reagents, 2.5 μl 25 μg/ml PNA Cy3-telomere probe, and 16.6 μl distilled water.) The slides were washed twice in washing solution I (70 ml formamide/1 ml 1M Tris Cl, pH 7.2/1 ml 10% BSA stock solution/28 ml distilled water), followed by three washes in washing solution II (15 ml 1M Tris Cl, pH 7.2/15 ml 1.5 M NaCl/120 μl Tween 20 (0.08% final)/120 ml distilled water). After dehydration in 70%, 90%, and 100% ethanol, samples were air dried for about 20 minutes in the dark. Pictures were taken at 2500 ms for Cy3 images and at 100 ms for DAPI images. Quantification of TFI was preformed using TFL-TELO V1.0, a telomere analysis program developed by P Landsdorp. To facilitate day to day comparison, one standard sample was photographed and analysed for each individual session. The correlation of TFI measured by Q-FISH and telomere length was confirmed using white blood cells of successive generations of telomerase deficient mice (data not shown).
RESULTS
Histology of the samples
In all 10 cases areas of high grade dysplasia showed minimal invasive growth—the earliest defined carcinoma stage during human colorectal carcinogenesis. Epithelial cells in areas of high grade dysplasia were characterised by loss of polarity, round, enlarged, and pleomorphic nuclei, increased rate of mitotic figures, and a cribriform architecture (fig 1A, B). To allow cell type specific analysis of telomere length (epithelial cells v cells of connective tissue), morphological criteria were used (fig 1C, D). Connective tissue consists of densely packed cells and vessels compared with epithelial cells in the adenoma and high grade dysplasia.
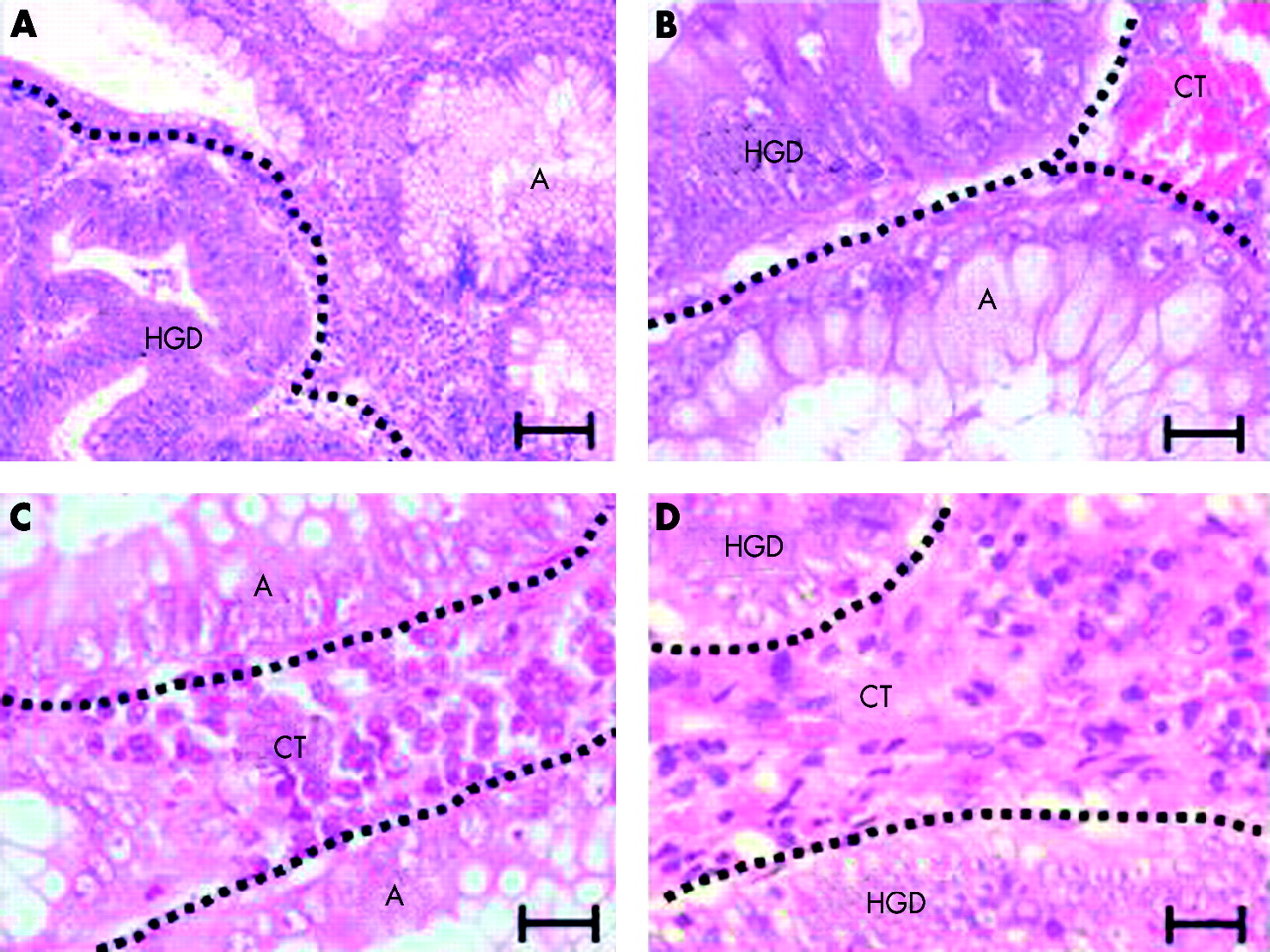
Morphological characteristics of the adenoma-carcinoma transition. Representative photographs of haematoxylin-eosin stained sections of adenoma (A) with high grade dysplasia (HGD) showing increased mitotic figures and a cribriform architecture. Epithelial cells in HGD are characterised by loss of polarity of the cells, and enlarged and pleomorphic nuclei. (A) Adenoma-carcinoma transition: the adenoma (A) on the right shows typical epithelial cells in a single cell layer whereas HGD on the left shows multiple layers of dysplastic epithelial cells (magnification bar 200 μm). (B) Adenoma-carcinoma transition: HGD at the top of the image includes epithelial cells with pleomorphic nuclei and mitotic figures, whereas the adenoma (A) at the bottom shows polarised epithelial cells and goblet cells. CT, connective tissue (magnification bar 50 μm). (C, D) Representative photographs of haematoxylin-eosin stained sections showing connective tissue cells (C) in the adenoma (A) and (D) in HGD (magnification bars 50 μm).
Critical short telomeres of epithelial cells characterise the adenoma-carcinoma transition
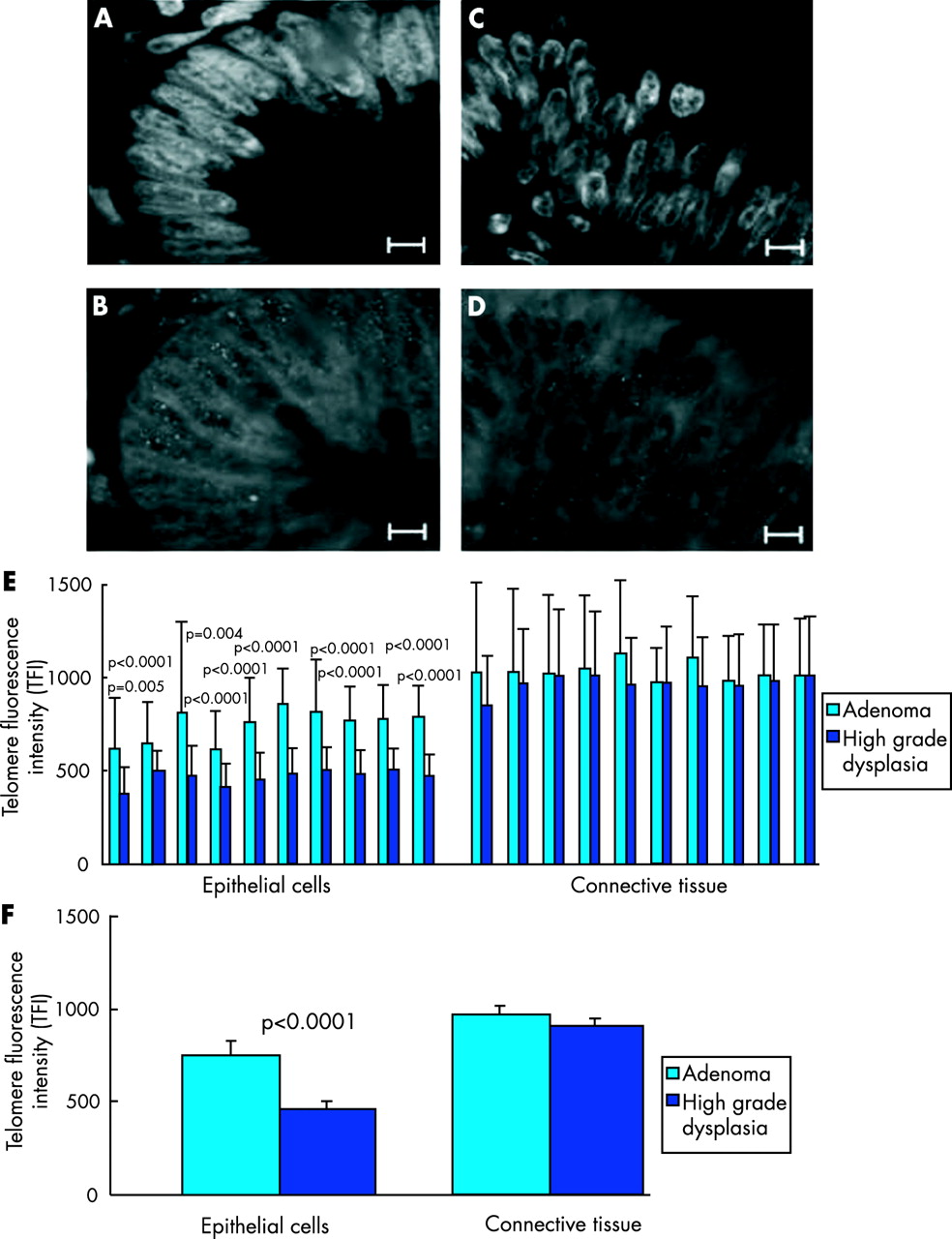
To test the hypothesis that critical short telomeres initiate cancer formation, Q-FISH analysis was carried out on 10 cases of sporadic colorectal cancer showing the adenoma-carcinoma transition. Quantification of TFI by Q-FISH analysis was performed on consecutive sections of the ones used for haematoxylin and eosin staining, thus allowing separate quantification of TFI in areas of high grade dysplasia and adenoma. To facilitate cell type specific analysis of telomere length, the samples used for telomere length analysis by Q-FISH were counterstained with DAPI showing the typical cell and nuclei morphology for epithelial cells and cells from connective tissue (fig 2A–D, fig 1). A minimum of 50 epithelial cell nuclei per sample were analysed for TFI using Q-FISH. Direct comparison of TFI between high grade dysplasia and surrounding adenoma revealed a significantly reduced telomere length in epithelial cells in areas of high grade dysplasia compared with the surrounding adenoma in each case investigated (fig 2E). Similarly, the whole mean value of all 10 samples showed a significant reduction in TFI for epithelial cells compared with high grade dysplasia with adenoma (mean TFI 463 units in high grade dysplasia compared with 741 units in the adenoma; p<0.0001) (fig 2F). In addition to reduced mean values, minimum TFI values of nuclei of epithelial cells from high grade dysplasia were significantly smaller compared with epithelial cells from surrounding adenoma (data not shown). Interestingly, the standard deviation of TFI in epithelial cells from high grade dysplasia was smaller compared with the standard deviation for TFI of epithelial cells in the surrounding adenoma. This finding could be due to the fact that telomeres in high grade dysplasia had reached a critical short length necessary for cell viability leading to death of cells with too short telomeres thus reducing the variability of the mean values of cells in high grade dysplasia compared with adenoma.

{kind=link}
{kind=link}
Critical telomere shortening of epithelial cells at the adenoma-carcinoma transition. (A) DAPI stained section of adenoma epithelial cell nuclei. (B) Adenoma epithelial cell nuclei stained with a telomere specific Cy3 labelled probe Cy 3-00-(CCCTAA)3. (C) DAPI stained section of epithelial cell nuclei of high grade dysplasia. (D) High grade dysplasia epithelial cell nuclei stained with a telomere specific Cy3-labelled probe Cy 3-00-(CCCTAA)3 (magnification bar in A–D 25 μm). (E) Histogram of telomere fluorescence intensity (TFI) comparing mean values between epithelial cells of the adenoma with epithelial cells of high grade dysplasia for each individual sample (left panel). In all 10 samples, TFI was significantly lower in epithelial cells at the high grade dysplasia stage compared with the surrounding adenoma. In contrast with epithelial cells, cells from connective tissue had a similar TFI in adenoma and high grade dysplasia of individual samples (right panel). (F) Total mean value for TFI of epithelial cells and cells of connective tissue comparing adenoma and high grade dysplasia of all samples.
Proposing that critical telomere shortening is mechanistically linked to the development of spontaneous colorectal cancer at the adenoma-carcinoma transition, we wished to test its cell type specificity at the same transition. One would expect that other cell types in the vicinity of the malignant tumour which are not themselves malignantly transformed would not show significant telomere shortening but that it is specific for epithelial cells which are the cell type of cancer origin. In line with this hypothesis, the TFI of cells of connective tissue was not different comparing adenoma and high grade dysplasia, in any of the 10 samples investigated (fig 2E). In line with these data, the total mean value of TFI of connective tissue cells in all 10 samples did not show a significant difference between adenoma and high grade dysplasia (fig 2F). In addition, there was no difference comparing the standard deviation of TFI of cells from connective tissue between high grade dysplasia and surrounding adenoma. Irrespective of the transformation stage, TFI was significantly weaker in epithelial cells compared with the cells of connective tissue, possibly because telomere shortening in the intestine affects predominantly epithelial cells which undergo a constantly high cell turnover whereas cells of connective tissue are relatively mitotically inactive. Taken together, our findings show that a significant reduction in telomere length comparing adenoma and high grade dysplasia is specific for epithelial cells but not present in cells from connective tissues at the adenoma-carcinoma transition during human colorectal cancer formation.
DISCUSSION
Our current study supports the telomere hypothesis of cancer initiation. We have shown that telomere shortening characterises the adenoma-carcinoma transition and that telomere shortening at this transition specifically affects epithelial cells, the cell type of origin of colorectal cancer. Previous studies on telomere length in human colorectal cancer have exclusively focused on advanced cancer stages.21–26 Our data provide the first direct evidence that telomeres are already shortened at the earliest detectable carcinoma stage—high grade dysplasia with minimal invasive growth—in human colorectal cancer formation. Given that these early carcinomas consist of only a couple of hundred to thousand cells, shortening of telomeres at this stage is probably not a consequence of cell division during cancer progression but suggests that carcinomas arise from cells with critical short telomeres. These data support the hypothesis that telomere dysfunction initiates cancer formation by induction of CIN. Additional support for this hypothesis derives from studies on the prevalence of CIN showing a sharp increase in aneuploidy at the adenoma-carcinoma transition.5–14 According to the telomere hypothesis of cancer initiation, CIN induced by critical telomere shortening and loss of telomere function triggers the genetic lesions necessary for cellular transformation. In line with the telomere hypothesis of cancer initiation, the rate of anaphase bridges—a sign of telomere dysfunction—sharply increases at this transition.13 Anaphase bridges are not highly specific for telomere dysfunction but together with our current data on critically short telomeres at the same time point (the adenoma-carcinoma transition) strongly support the telomere hypothesis of induction of CIN and cancer initiation.
How does activation of telomerase that occurs in over 80% of human colorectal cancers22,27,28 support the hypothesis that telomere shortening initiates cancer by induction of CIN? A possible explanation has come from studies in telomerase deficient mice (mTERC−/−) showing that telomere shortening has an ambivalent role in carcinogenesis. In this mouse model telomere shortening increases cancer initiation by induction of CIN18,19,20 but at the same time inhibits cancer progression by p53 dependent induction of cell cycle arrest and apoptosis.19,29,30 In human colorectal cancer formation, activation of telomerase has been described at the adenoma-carcinoma transition,22,27,28 suggesting that it is a necessary step for cancer progression taking place in parallel or shortly after induction of carcinoma formation. According to the telomere hypothesis of cancer initiation, we propose a model suggesting that carcinomas arise from chromosomal unstable epithelial cells with critical short telomeres which have lost DNA damage responses. Subsequently or simultaneously, a variety of cofactors are necessary for cancer progression, including activation of telomerase to stabilise telomeres and alleviate CIN to a level allowing further cell divisions and cell survival.