Article Text

Abstract
Objective Vitamin D and the vitamin D receptor (VDR) appear to be important immunological regulators of inflammatory bowel diseases (IBD). Defective autophagy has also been implicated in IBD, where interestingly, polymorphisms of genes such as ATG16L1 have been associated with increased risk. Although vitamin D, the microbiome and autophagy are all involved in pathogenesis of IBD, it remains unclear whether these processes are related or function independently.
Design We investigated the effects and mechanisms of intestinal epithelial VDR in healthy and inflamed states using cell culture models, a conditional VDR knockout mouse model (VDRΔIEC), colitis models and human samples.
Results Absence of intestinal epithelial VDR affects microbial assemblage and increases susceptibility to dextran sulfate sodium-induced colitis. Intestinal epithelial VDR downregulates expressions of ATG16L1 and lysozyme, and impairs antimicrobial function of Paneth cells. Gain and loss-of-function assays showed that VDR levels regulate ATG16L1 and lysozyme at the transcriptional and translational levels. Moreover, low levels of intestinal epithelial VDR correlated with reduced ATG16L1 and representation by intestinal Bacteroides in patients with IBD. Administration of the butyrate (a fermentation product of gut microbes) increases intestinal VDR expression and suppresses inflammation in a colitis model.
Conclusions Our study demonstrates fundamental relationship between VDR, autophagy and gut microbial assemblage that is essential for maintaining intestinal homeostasis, but also in contributing to the pathophysiology of IBD. These insights can be leveraged to define therapeutic targets for restoring VDR expression and function.
- Inflammatory Bowel Disease
- Vitamin D Receptor Gene
- Epithelial Cells
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Vitamin D and the vitamin D receptor (VDR) appear to be important immunological regulators of inflammatory bowel diseases (IBD).
Low vitamin D status has been observed in patients with IBD.
A north-south gradient in rates of Crohn's disease (CD) suggests that vitamin D deficiency may be an environmental trigger contributing to the pathogenesis of IBD.
Moreover, polymorphisms in the VDR gene are associated with susceptibility to IBD. VDR expression is significantly decreased in patients with IBD.
Bacteria regulate intestinal VDR expression in both gnotobiotic and bacterial-colitis models. Under conditions of intestinal inflammation, VDR negatively regulates bacteria-induced proinflammatory NF-κB activity.
What are the new findings?
Our study for the first time links dysbiosis, innate immune functions (Paneth cells) and genetic susceptibility through intestinal epithelial VDR.
Our data provide insights by specifically investigating how intestinal epithelial VDR regulates autophagy and Paneth cells through the autophagy gene ATG16L1, thus changing the microbiome profile.
We further show that low levels of VDR correlate with decreased ATG16L1 in the intestine of patients with IBD and in an experimental colitis model.
Gain and loss-of-function assays show that VDR transcriptionally regulates ATG16L1, thus decreasing lysozyme.
Finally, administration of the bacterial product butyrate increases intestinal VDR and ATG16L1 expression and suppresses inflammation in an experimental colitis model.
Our study fills an existing gap by characterising the precise role of intestinal epithelial VDR in regulating intestinal homeostasis through alterations in intestinal autophagy and the microbiome. It also brings up the possibility that microbial natural products can be used therapeutically to restore VDR-dependent functions in patients with IBD.
How might it impact on clinical practice in the foreseeable future?
Our studies demonstrate a fundamental relationship between vitamin D, VDR, Paneth cell function, gut microbiota and genetic susceptibility genes (ATG16L1) that is essential for the maintenance of intestinal homeostasis, but also for the development of chronic states of mucosal inflammation. This knowledge can be immediately used to develop intestinal VDR as a clinical biomarker for identifying patients who might benefit from currently available interventions, as well as for the eventual development of novel strategies for the prevention and treatment of human IBD.
Introduction
Vitamin D deficiency is a critical factor in the pathology of inflammatory bowel diseases (IBD), colon cancer, autoimmune diseases and other diseases.1–7 Low vitamin D status has been observed in patients with IBD.8–10 A north-south gradient in rates of Crohn's disease (CD) suggests that vitamin D deficiency may be an environmental trigger contributing to the pathogenesis of IBD.9 Vitamin D also influences the course and severity of IBD.9 The vitamin D receptor (VDR), a nuclear receptor,11 mediates most known functions of 1,25-dihydroxyvitamin D3 (vitamin D3), the active form of vitamin D. VDR and vitamin D3 are recognised as key players in calcium homeostasis and in electrolyte and blood pressure regulation.12 More recent evidence demonstrates that vitamin D3 is an immunoregulatory hormone that modulates the innate and adaptive immune system.13–15 Vitamin D/VDR appears to be important immunological regulators of IBD.16 ,17 Moreover, polymorphisms in the VDR gene are associated with increased susceptibility to IBD.18–22 VDR expression is significantly decreased in patients with IBD.8 ,23 Although it is possible that decreased VDR is a consequence of chronic inflammation in IBD, in experimental murine models of colitis, VDR knockout (KO) mice spontaneously develop colitis.24 Whole-body VDR/IL-10 double KO mice develop a more severe colitis involving the entire intestinal tract.24 Mice lacking VDR activate the proinflammatory NF-κB pathway and are susceptible to Salmonella colitis and chemical-induced colitis.25 ,26 These findings suggest a critical role of VDR in maintaining intestinal homeostasis and possibly in causing/contributing to human IBD.
VDR also functions as a transcription factor.27 Target genes of VDR include antimicrobial peptide (AMPs) cathelicidin antimicrobial peptide 28 ,29 (also called LL-37), β-defensin29 and the 1,25(OH)2D3-regulated VDR-specific, Cyp24 hydroxylase gene. Indeed, ∼3% of the mouse and human genomes are regulated directly or indirectly by the vitamin D endocrine system, further supporting the possibility of widespread effects of vitamin D and VDR in disease mechanisms.30–32 For example, epidemiological and experimental studies have indicated a protective action of vitamin D against colorectal cancer and upper gastrointestinal cancers.33–40 Upregulation of VDR leads to induction of AMPs and the killing of intracellular Mycobacterium tuberculosis in human macrophages.41 Paneth cells are specialised intestinal epithelial cells located at the bottom of ileal crypts. The granules of Paneth cells contain AMPs—α-defensins, lysozyme and secretory phospholipase A2.42–44 Paneth cells are a major source of monocyte chemotactic protein 1(MCP-1)45 and also produce the cytokine IL-17.46 A recent study demonstrated Paneth cells as a site of origin for intestinal inflammation.47 Thus, Paneth cells play a key role in innate immune responses and in shaping the gut microbiota.48 However, VDR regulation of Paneth cell function is not known.
Autophagy is a highly conserved process that is involved in intracellular homeostasis through the degradation and recycling of cytosolic contents and organelles, as well as in promoting the removal of intracellular microbes and immunity against infection.49 ,50 Interestingly, three IBD susceptibility genes, IRGM, Nod2 and ATG16L1, are involved in autophagy.51–53 Deficits in the autophagy pathway can impair Paneth cell function in patients with CD.54 ,55 Autophagy can affect the biosynthesis or quality control of the lysosomal pathway in Paneth cell granules. Paneth cells with fewer granules might be a result of Paneth cell exhaustion, compensating for changes elsewhere in the epithelium.54 Expression in CD-like mutant Nod2 mouse alleles affects Paneth cells by reducing Nod2-dependent production of α-defensins in Paneth cells.56 Nod2 and ATG16L1 are in a common bacterial-sensing pathway that promotes bacterial antigen presentation. Nod2 recruits ATG16L1 to the plasma membrane at the bacterial entry site.57 Moreover, ATG16L1 may have unique protective functions, including AMP release in Paneth cells and the negative regulation of proinflammatory cytokine production.58 ,59 Although vitamin D and autophagy are all involved in pathogenesis of IBD,60 it remains unclear whether these processes are related or function independently. Several studies61 ,62 have identified vitamin D as a potent stimulator of autophagy in M. tuberculosis infection and HIV infection. However, the crosstalk among VDR, autophagy and bacteria in the gut remains unknown.
We have been investigating VDR63 ,64 and bacterial inflammation.25 ,63–66 We found that, on the one hand, VDRs negatively regulate bacteria-induced NF-κB activity in intestinal inflammation.63 Lack of VDR leads to a reduction of IκBα, an endogenous inhibitor of NF-κB activity. On the other hand, bacteria regulate intestinal VDR expression in gnotobiotic and bacterial-colitis models.63 Recent studies have also shown alternate bacterial profiles in VDR KO mice. VDR may regulate the gut microbes and probably contribute to maintenance of physiological host–microbe relationships. This could occur through several unique mechanisms that include NF-κB and autophagy. In the current study, we hypothesise that the intestinal epithelial VDR is a determinant of IBD risk through its actions on the autophagy gene (ATG16L1), thus determining states of Paneth cells and microbial assembly in intestinal homeostasis. We investigated mechanisms of intestinal epithelial VDR in healthy and inflamed states using a conditional KO mouse model. We show that mice lacking VDR have increased bacterial loads in intestinal mucosa. The number of Paneth cells is decreased in the ileum of VDR−/− mice compared with control mice. We report that VDR levels correlated with levels of autophagy markers in vitro and in vivo. We further show that administration of the bacterial product butyrate increases intestinal VDR expression and suppresses inflammation in an experimental colitis model. Hence, our study fills an existing gap by characterising the precise role of intestinal VDR in regulating intestinal homeostasis through changing the function of intestinal autophagy.
Materials and methods
Human tissue samples
This study was performed in accordance with approval from the University of Rochester Ethics Committee (RSRB00037178). Colorectal tissue samples were obtained with informed consent from the sigmoid colon of 52 patients (51–83 years old) exhibiting no apparent intestinal pathology and from the normal mucosa (ie, >10 cm colitis margin) of 30 patients undergoing anterior resection (44–85 years old).
Animals
VDRloxP/loxP mice were originally developed by Dr Geert Carmeliet.67 VDRΔIEC mice were obtained by crossing the VDRloxP/loxP mice with villin-cre mice (Jackson Laboratory, 004586). Experiments were performed on 2–3 months old mice. Mice were provided with water ad libitum and maintained in a 12 h dark/light cycle. IL10−/− mice were purchase from Jackson Laboratory (002251). All work with animals was approved by the Rush University Animal Resources Committee.
Induction of colitis
Mice were administered 5% dextran sulfate sodium (DSS) (MW = 40–50 kDa; USB Corp. Cleveland, OH) dissolved in filter-purified and sterilised water ad libitum for the experimental period. Animals were weighed daily. At day 7 after DSS administration, mice were sacrificed under anaesthesia. Severity of colitis was quantified by a disease activity index, determined by weight loss, faecal blood and diarrhoea.68
Butyrate-treated mice model
Eight IL10−/− mice were divided into control and butyrate groups. Butyrate group mice were given 2% sodium butyrate (Sigma, St. Louis, Missouri, USA) water ad libitum for 3 weeks. The untreated control group was maintained on filtered water throughout the experiment.
Co-housing experiment
Two-to-three-month-old female VDRloxP/loxP and VDRΔIEC were co-housed in new cages according to previously published methods.69 One cage contained three VDRloxP/loxP and two VDRΔIEC, another one contained two mice each. The mice were fed with the same food and water. After 4 weeks of co-housing, 5% DSS dissolved in filter-purified water was administered to the mice. Animals were weighed daily. At day 7 after DSS administration, mice were sacrificed under anaesthesia.
Cell culture
Mouse embryonic fibroblasts (MEFs), human embryonic intestine INT 407, HCT116 cells and human colorectal adenocarcinoma SKCO-15 cells were grown in high glucose Dulbecco's modified Eagle medium (DMEM) (Hyclone, SH30243.01) containing 10% (v/v) fetal bovine serum (GEMINI, 900–108), 50 μg/mL streptomycin and 50 U/mL penicillin (Mediatech, Inc., 30-002CI). INT 407 cells were grown with 1% non-essential amino acids (Mediatech, Inc., 25-025CI).
VDR knockdown of SKCO 15 with shRNA using cells retroviral GFP vector
SKCO-15 cells were transfected (Lipofectamine 2000, Invitrogen Corp.) with VDR shRNA retroviral GFP vector (OriGene, TG320568) plasmids targeting human VDR or control, scrambled shRNA on the same vector background (Origene) according to the manufacturer's instructions. Briefly, SKCO-15 cells were seeded on day 1 in a six-well plate with complete medium and incubated overnight. Medium was replaced on day 2 with fresh complete medium containing 5 μg/mL puromycin dihydrochloride (GIBCO A11138-02) for 6 days. The transduced cells were sorted by GFP detection using flow cytometry and cultured in complete medium containing 10 μg/mL puromycin dihydrochloride for 13 passages.
Vitamin D-responsive element transcriptional activity
Cells were grown in triplicate and transfected with Cignal Vitamin D Reporter (luc) Kit (SABiosciences, Frederick, Maryland, USA) using Surefect reagent (SABiosciences, Frederick, MD). The plasmid for the VDR reporter was a mixture of an inducible vitamin D-responsive firefly luciferase construct and a constitutively expressing Renilla luciferase construct (40:1). The negative control was a mixture of a non-inducible firefly luciferase construct and a constitutively expressing Renilla luciferase construct (40:1). After transfection for 24 h, some cells were treated with 10 mM butyrate for 30 h. Luciferase activity was determined using the Dual Luciferase Reporter Assay System (Promega) with a TD- 20/20 luminometer (Turner Designs, Sunnyvale, California, USA).
Western blot analysis and antibodies
Mouse ileal epithelial cells were collected by scraping from mouse ileum as previously described.65 Briefly, mouse epithelial cells were lysed in lysis buffer (1% Triton X-100 (Sigma-Aldrich, X100), 150 mM NaCl (J.T.Baker 3624-19), 10 mM Tris (Fisher Scientific, BP152-5) pH 7.4, 1 mM EDTA (Fisher Scientific, BP120-1), 1 mM EGTA (Sigma-Aldrich, E3889) pH 8.0, 0.2 mM sodium ortho-vanadate (Sigma-Aldrich, S6508) and protease inhibitor cocktail (Roche Diagnostics, 118367001)). Cultured cells were rinsed twice in ice-cold Hanks' balanced salt solution (Sigma-Aldrich, H1387), lysed in protein loading buffer (50 mM Tris, pH 6.8, 100 mM dithiothreitol (Amresco, 0281), 2% SDS (Sigma-Aldrich, L3771), 0.1% bromophenol blue (IBI Scientific, IB74040) and 10% glycerol (Sigma-Aldrich, G5516)) and sonicated (Branson Sonifier, 250). Equal amount of protein was separated by SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose (Bio-rad, 162-0112) and immunoblotted with primary antibodies: VDR (Santa Cruz, sc13133), Beclin-1 (Santa Cruz, sc10086), Villin (Santa Cruz, sc7672), LC3B (Cell Signal Technology Inc., 2775), ATG16L1 (Abgent, AP18176), p62 (Abgent, AP2183B) or β-actin (Sigma-Aldrich, A1978) antibodies and visualised by ECL chemiluminescence (Thermo Scientific, 32106).
Histology of Intestine
Intestines were harvested, fixed in 10% formalin (pH 7.4), processed and paraffin embedded. Sections (5 μm) were stained with H&E. For immunostaining, antigens were retrieved by 10 min boiling in 10 mM citrate (pH 6.0). The slides were stained with antibodies as previously described.65 Blinded histological inflammatory scores were performed by a validated scoring system by a trained pathologist.70
Immunofluorescence
Ileal tissues from the distal portion of the ileum were freshly isolated and paraffin-embedded after fixation with 10% neutral buffered formalin. Immunofluorescence was performed on paraffin-embedded sections (5 μm). After preparation of the slides as described previously,71 tissue samples were incubated with antilysozyme (Santa Cruz, sc27958) at 4°C overnight. Samples were then incubated with sheep antigoat Alexa Fluor 594 (Life Technologies, A11058) and DAPI (Life Technologies, D1306) for 1 h at room temperature. Tissues were mounted with SlowFade (Life Technologies, s2828), followed by a coverslip, and the edges were sealed to prevent drying. Specimens were examined with Zeiss laser scanning microscope (LSM) 710.
Fluorescence in situ hybridisation
Fluorescent in situ hybridisation (FISH) was performed using antisense ssDNA probes targeting the bacterial 16S rRNA. Bfra602 probe (5′-GAGCCGCAAACTTTCACAA-3′) for Bacteroides fragilis group.72 Prior to performing the FISH assay, 5 μm tissue sections were baked overnight at 55°C. Tissue sections were deparaffinised in xylene, dehydrated with 100% ethanol, air dried, incubated in 0.2 M HCl for 20 min and heated in 1 mM sodium thiocyanate at 80°C for 10 min. Samples were pepsin digested (4% pepsin in 0.01N HCl) for 20 min at 37°C, washed on slides in wash buffer (0.3 M NaCl, 0.03 M sodium citrate, pH 7 and 0. 1% SDS) and fixed on slides in 10% buffered formalin for 15 min and hybridised with the probes at 5 ng/μL concentration each for 5 min at 96°C in hybridisation buffer (0.9 M NaCl, 30% formamide, 20 mMTris-HCl (pH 7.4) and 0.01% sodium dodecyl sulfate (SDS)) and incubated at 37°C overnight. Slides were washed four times for 5 min each at 45°C in wash buffer. For visualisation of the epithelial cell nuclei, the slides were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI)/antifade solution. The slides were examined with Zeiss laser scanning microscope (LSM) 710.
Lysotracker staining
Lysotracker-red is a basic cell-permeable probe that accumulates in acidic vesicles. It is widely used to reflect lysosomal activity in live cells.61 ,73 ,74 Lysotracker staining was performed following the manufacturer protocol (Lonza Walkersville, Inc.). MEF and INT 407 cells were grown in the Lab-Tek Chambered Cover glass System (Thermo Scientific, 154526), and the cells were then incubated with 100 nM LysoTracker Red lysosomal Probe (Lonza Walkersville, Inc., PA3015) in cell growth medium at 37°C for 60 min. After washing with HBBS, the cells were detected by fluorescence microscopy (AMG, EVOS fl).
Paneth cells counting
Paneth cells in mouse ileal cells were counted after antilysozyme immunofluorescence staining. The patterns of lysozyme expression in Paneth cells were classified into four categories: normal (D0), disordered (D1), depleted (D2) and diffuse (D3) according to published methods.59
Real-time quantitative PCR
Total RNA was extracted from mouse ileal epithelial cells or cultured cells using TRIzol reagent (Life Technologies, 15596-02). RNA was first reverse-transcribed into cDNA with the iScript cDNA synthesis kit (Bio-Rad, 170-8891) according to the manufacturer's manual. The RT-cDNA reaction products were subjected to quantitative real-time PCR using CTFX 96 Real-time system (Bio-Rad, C1000) and SYBR green supermix (Bio-Rad, 172–5124) according to the manufacturer's directions. All expression levels were normalised to β-actin levels of the same sample. Per cent expression was calculated as the ratio of the normalised value of each sample to that of the corresponding untreated control cells. All real-time PCR reactions were performed in triplicate. Optimal primer sequences were designed using Primer-BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) or were obtained from Primer Bank (http://pga.mgh.harvard.edu/primerbank/) primer pairs listed (table 1).
Real-time PCR primers
Real-time PCR measurement of bacterial DNA
DNA was extracted from colonic tissues.75 16S rDNA PCR reactions were performed with the following primers: Universal bacteria76 (forward: 5′-TCCTACGGGAGGCAGCAGT-3′; reward: 5′-GGACTACCAGGGTATCTAATCCTGTT-3′), Escherichia coli (forward: 5′-CCTACGGGAGGCAGCAGT-3′; reward: 5′-CGTTTACGGCGTGGACTAC-3′), B. fragilis (forward: 5′-GGCGCACGGGTG-AGTAACA-3′; reward: 5′-CAATATTCCTCACTGCTGC-3′) and Butyrivibrio fibrisolvens (forward: 5′-CTAACACATGCAAGTCGAACG-3′; reward: 5′-CCGTGTCTCAGTCCCAATG-3′), primers specific to 18S rRNA77 (forward: 5′-AGGGGAGAGCGGGTAAGAGA-3′; reward: 5′-ggacaggactaggcg-gaaca-3′) were used as an endogenous control to normalise loading between samples. The relative amount of 16S rDNA in each sample was estimated using the ΔΔCT.
Mucosa microbial and faecal 454 pyrosequencing
The tubes for microbial sampling were autoclaved and then irradiated with ultraviolet light to destroy the environmental bacterial DNA. The mice were then anaesthetised and dissected. Faecal were isolated freshly from the gut and placed into the specially prepared tubes, as described in our previously published papers.78 ,79 The samples were kept at low temperature with dry ice and mailed to Research and Testing Laboratory, Lubbock, TX, for 454 pyrosequencing. The V4-V6 region of the samples was amplified in Research and Testing Laboratory, Lubbock, TX, for pyrosequencing using a forward and reverse fusion primer. The sequences were denoised, subjected to quality checking. Taxonomic identifications were assigned by queries against National Center for Biotechnology Information. Differences in microbial communities between VDRloxP/loxP and VDRΔIEC groups were analysed, as we did in previous studies.78 ,79 Briefly, principal coordinates analysis (PCoA) of unweighted UniFrac distances plots were plotted using quantitative insights into microbial ecology (QIIME).80 To determine differences in microbiota composition between the animal groups, the analysis of similarities (ANOSIM) function in the statistical software package PRIMER 6 (PRIMER-E Ltd., Lutton, UK) was used on the unweighted UniFrac distance matrixes.
Chromatin immunoprecipitation (CHIP) assay
Binding of VDR to the ATG16L1 promoter was investigated using the ChIP assay as described previously.25 Briefly, MEF cells were treated with 1% formaldehyde for 10 min at 37°C. Cells were washed twice in ice-cold phosphate buffered saline containing protease inhibitor cocktail tablets (Roche). Cells were scraped into conical tubes, pelleted and lysed in SDS Lysis Buffer. The lysate was sonicated to shear DNA into fragments of 200–1000 bp (four cycles of 10 s sonication, 10 s pausing, Branson Sonifier 250, USA). The chromatin samples were precleared with salmon sperm DNA–bovine serum albumin-sepharose beads, then incubated overnight at 4°C with VDR antibody (Santa Cruz Biotechnology). Immune complexes were precipitated with salmon sperm DNA–bovine serum albumin-sepharose beads. DNA was prepared by treatment with proteinase K, extraction with phenol and chloroform, and ethanol precipitation. Searching mouse ATG16L1 gene, we found a similar sequence as the vitamin D response element (VDRE) sequence ‘(G/A)G(G/T)TCA’. We then designed primers for ChIP. PCR was performed using the following promoter-specific primers: ATG16L1 forward, 5′-GGTTCCGTTCTTGTTTCT- 3′; reverse, 5′-TCAAGTTGT-CTCCAAGATTAT-3′.
Statistical analysis
Data are expressed as mean±SD. Differences between two samples were analysed by Student's t test. Differences among three or more groups were analysed using ANOVA with GraphPad Prism 5. p Values of 0.05 or less were considered statistically significant.
Results
Established intestinal VDRΔIEC model
We have established a VDRΔIEC model using the VDRloxP/loxP mouse strain.67 ,81 As shown in figure 1A and B, there was no detectable VDR expression in intestinal epithelial cells of VDRΔIEC mice by immunohistochemistry or by Western blot analysis. These data confirm that the intestinal VDR KO is established. The VDRΔIEC model allows us to (1) focus on intestinal epithelial VDR and clearly define the mechanisms by which intestinal epithelial VDR regulates intestinal homeostasis, (2) study a well-controlled system without having to feed the mice a special calcium-enriched diet and (3) avoid other non-digestive disorders found in total VDR−/− mice.

Established intestinal epithelial cell vitamin D receptor (VDR) knockout (KO) (VDRΔIEC) mice and their bacterial profile. (A) VDR immunohistochemistry staining in intestine. (B) Western blotting of intestinal VDR. Villin is a marker for IECs. (C) Real-time PCR of bacterial universal 16srDNA and 16 s rDNA for Escherichia coli, Bacteroides fragilis and Butyrivibrio fibrisolvens. Primers specific to 18S rRNA were used as an endogenous control to normalise loading between samples. The relative amount of 16S rRNA in each sample was estimated using the ΔΔCT method (n=3,* p<0.05). (D) Principal coordinates analysis (PCoA) of unweighted UniFrac distances of 16 S rRNA genes. Analysis for VDRloxP/loxP (blue) and VDRΔIEC (red) mice. The results indicate that faecal microbial communities differ in VDRΔIEC mice compared with VDRloxP/loxP. (E) Bacterial community of faecal samples from VDRΔIEC and VDRloxP/loxP mice using 454 16S rRNA sequencing data (n=4/group). (F) Representative fluorescence in situ hybridization staining for B. fragilis (red) in VDRΔIEC and VDRloxP/loxP control mice. Blue, DAPI.
Absence of intestinal epithelial VDR leads to ecological change in bacterial profiles
Interestingly, bacterial abundance is significantly changed in VDRΔIEC mice compared with the VDRloxP/loxP mice without any treatment, demonstrating increased E. coli and Bacteroides and decreased butyrate-producing bacteria (Butyrivibrio) (figure 1C). Our 454 16S rRNA sequencing data also show the altered bacterial profile in the VDRΔIEC mice. PCoA indicated that faecal microbial communities differ in VDRΔIEC mice compared with VDRloxP/loxP (figure 1D). The relative abundance of bacteria was shifted in VDRΔIEC mice compared with VDRloxP/loxP (figure 1E). These data are consistent with our 454 16S rRNA sequencing of cecal and proximal colon samples that revealed different taxa abundances with a significant increase in the proportion of Bacteroides in whole-body VDR KO mice relative to wild-type (WT) mice (data not shown). These results are also consistent with a study that showed commensal Bacteroides species induce colitis.82 B. fragilis, a common human commensal microbiota, has been associated with IBD.83–85 To examine the distribution and abundance of bacteria, we performed FISH staining for B. fragilis. Our data show enhanced B. fragilis in the VDRΔIEC mice compared with the VDRloxP/loxP mice (figure 1F).
VDRΔIEC mice are susceptible to chemical injury
To investigate the biological effects of intestinal epithelial VDR in response to injury, we used a DSS-colitis model.86 Our data showed that VDRΔIEC mice were susceptible to DSS-mediated inflammation than VDRloxP/loxP mice (figure 2). The VDRΔIEC mice had significant loss of body weight after DSS treatment for 7 days (figure 2A). The caecum length was significantly reduced in the VDRΔIEC mice with DSS compared with VDRΔIEC mice without DSS, whereas no significant change was found between VDRloxP/loxP mice with or without DSS (figure 2B). In VDRΔIEC mice with DSS, faecal blood was more obvious, stools were less formed and more weight was lost. Accordingly, the Disease Activity Index was significantly increased compared with the VDRloxP/loxP group (p<0.05). (figure 2C). The H&E staining data in figure 2D showed more severe intestinal inflammation in VDRΔIEC mice with DSS compared with the VDRloxP/loxP mice. Next, the level of intestinal inflammation was quantified histologically in a blinded fashion by a trained pathologist. The inflammation score in the DSS-treated VDRΔIEC mice was significantly higher than the DSS-treated VDRloxP/loxP mice (figure 2E). Since the microbiota may play a role in the vulnerability of the VDRΔIEC mice to DSS, we examined the transmissibility of the phenotype by performing a co-housing experiment and then challenging the mice with DSS. We found that the co-housing increased disease activity index of VDRloxP-loxP mice to a level similar to that seen with the VDRΔIEC mice (figure 2F compared with 2C). The co-housing data suggest that the absence of intestinal epithelial VDR confers a transmissible risk for DSS-induced colitis. Taken together, our data indicate that intestinal VDR contributes to host protection against inflammation.

VDRΔIEC mice have worse outcomes with dextran sulfate sodium (DSS)-induced colitis. (A) Relative body weight changes in mice with or without DSS. (B) Caecum shorten was found in VDRΔIEC mice with colitis. (C) Disease Activity Index of colon from mice with or without DSS treatment. (D) Representative H&E histology of colon from mice with or without DSS treatment. (E) Inflammatory scores of the mouse intestine with or without DSS. VDRloxP/loxP and VDRΔIEC mice were administrated with DSS. n=5/group, *p<0.05. (F) Inflammatory scores of intestine after co-housing VDRloxP/loxP and VDRΔIEC mice. Two-to-three-month-old female VDRloxP/loxP and VDRΔIEC were co-housed in new cages at 3:2 or 2:2 mice. After 4 weeks of co-housing, the mice were administered with 5% DSS for 7 days to induce colitis. The dysbiosis associated with VDR-deficient mice appears to confer risk of chemically induced colitis. VDRloxP/loxP mice co-housed with VDRΔIEC mice developed the similar inflammation as the VDRΔIEC mice.
Absence of intestinal epithelial VDR leads to abnormal Paneth cells
Paneth cells are specialised intestinal epithelial cells that secrete AMPs,42–44 sense commensal bacteria and maintain homeostasis at the intestinal–microbial interface.48 We counted the number of Paneth cells using a previously reported method to stain lysozyme.59 Abnormal Paneth cells were grouped as D1 (disordered), D2 (depleted) and D3 (diffuse) (figure 3A). We found fewer than normal Paneth cells in the VDRΔIEC mice ileum than in VDRloxP/loxP mice (figure 3B). The abnormal Paneth cells D1–D3 increased in the VDRΔIEC mice (figure 3C). Abnormal Paneth cells lead to decreased lysozyme (Lyz1 and Lyz2) at the mRNA level (figure 3D).

Vitamin D receptor (VDR) affects patterns of Paneth cells in VDRΔIEC mice. (A) Representative images showed patterns of Paneth cells. D0, normal (yellow circle); D1, disordered (green circle); D2, depleted (yellow circle); D3, diffuse (yellow circle). (B) Representative images of indirect immunofluorescence of sections stained for lysozyme (red) in ileal crypts of VDRΔIEC and VDRloxP/loxP mice. (C) Percentage of Paneth cells displayed normal and abnormal (D1 to D3) patterns of lysozyme expression (n=10/group, *p<0.05). (D) Decreased lysozyme (lyz) in VDRΔIEC mice (n=3/group, *p<0.05).
Intestinal epithelial deletion of VDR downregulates ATG16L1 and autophagy that can impair Paneth cell function
Deficits in the autophagy pathway can impair Paneth cell function. Autophagy plays an essential role in innate immunity.49 The IBD susceptibility gene ATG16L1 is involved in autophagy and contributes to inflammation and dysbiosis.87 Our data showed that deletion of intestinal epithelial VDR leads to a significant reduction of ATG16L1 protein in vivo (figure 4A). The protein level of lysozyme, another component of the autophagy pathway, was also significantly lower in VDRΔIEC mice (figure 4A). We found that mRNA levels of ATG16L1 were lower in VDRΔIEC mice compared with the control VDRloxP/loxP mice (figure 4B). P62 is a signalling adaptor that accumulates in autophagy-deficient mice.88 ,89 We also found p62 protein increased in VDRΔIEC mice (figure 4C).
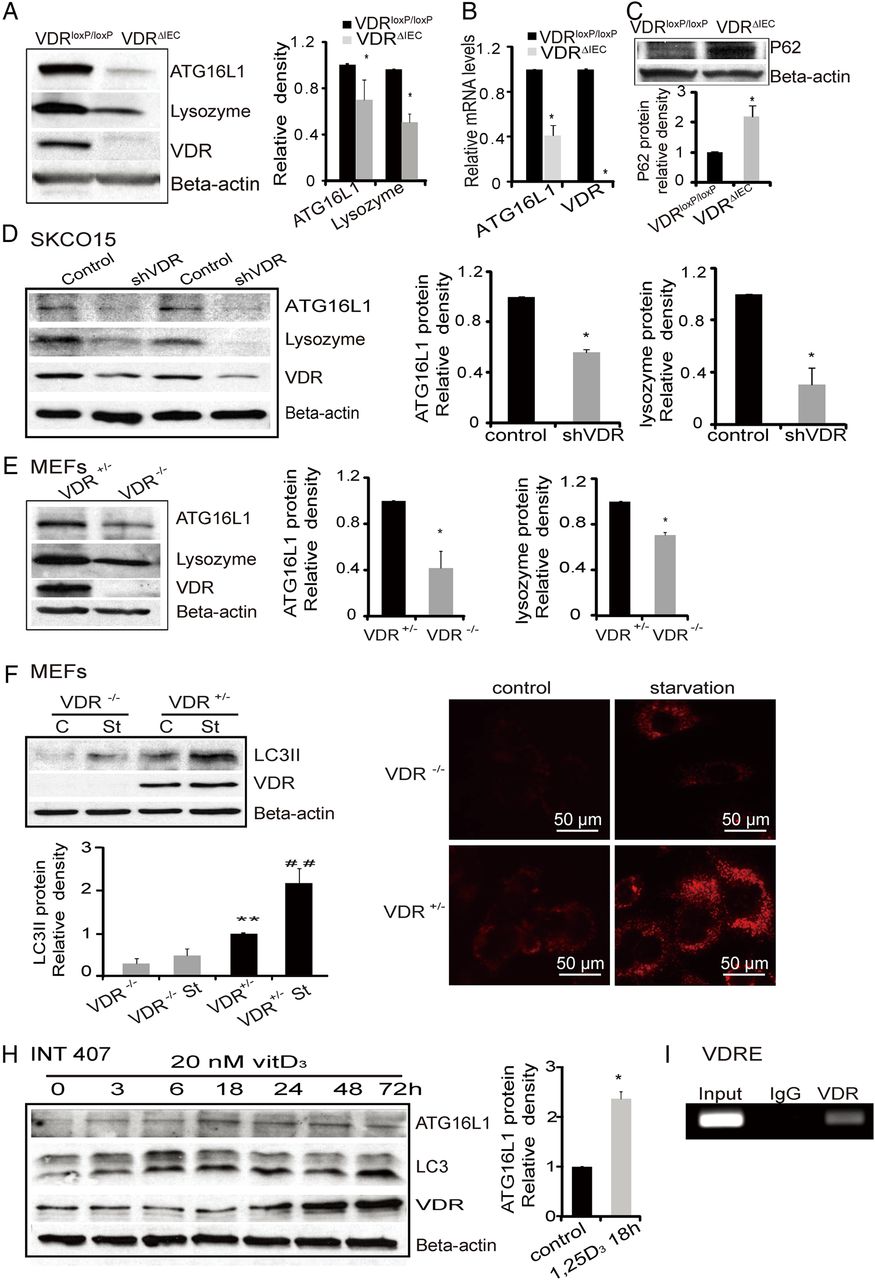
Vitamin D receptor (VDR) regulation of the expression levels of autophagy-related genes. (A) Levels of ATG16L1 protein and lysozyme in VDRΔIEC and VDRloxP/loxP mice. The relative intensity of ATG16L1 and lysozyme. (n=3/group. *p<0.05). (B) ATG16L1 mRNA level decreased in VDRΔIEC (n=3, *p<0.05). (C) Levels of P62 protein in VDRΔIEC and VDRloxP/loxP mice. The relative intensity of P62 (n=3 /group; *p<0.05). (D) ATG16L1 and lysozyme were decreased in intestinal epithelial cells with knockdown VDR by SiRNA VDR. (E) Lacking of VDR induced the reductions of autophagy marks (ATG16L1 and lysozyme) in mouse embryonic fibroblast (MEF) cells. (F) Expression of LC3 II in VDR−/− and VDR+/− MEF cells. MEF cells were cultured to 80% confluence. Incubated in fetal bovine serum (FBS) free E-MEM for 2 h as starvation (st). Cell lysates were immunoblotted with antibodies against LC3B and β-actin. (G) Representative images of lysotracker staining in MEF VDR−/− and VDR+/− cells. MEF cells were grown in the Lab-Tek Chambered coverglass System, incubated in FBS-free E-MEM for 2 h as starvation, and incubated with 100 nM LysoTracker red lysosomal probe for 60 min. After washing with HBBS, the cells were detected under fluorescence microscopy (AMG, EVOS fl). (H) ATG16L1 was increased with the other autophagy markers (LC3, ATG16L1) in intestinal INT407 cells with enhanced VDR by vitamin D treatment. (I) Regions encompassing putative vitamin D-response element amplified by PCR in ChIP assays in vitro.
VDR KO decreases ATG16L1 and LC3B proteins in SKCO15 and MEF cells
The ATG genes control autophagosome formation through ATG12-ATG5 and microtubule-associated protein1 light chain 3 (LC3-II or ATG8-II) complexes. In a loss-of-function experimental model, we further determined the effects of VDR on autophagy. We knocked down VDR protein with siRNA in human colon adenocarcinoma-derived epithelial SKCO15 cells. We found that VDR knockdown led to significantly decreased ATG16L1 and lysozyme at the protein levels (figure 4D).
We used VDR−/− and VDR+/− mouse embryonic fibroblast (MEF) cells to test whether one allele of the VDR gene was sufficient to re-establish a normal autophagy phenotype and found that complete lack of VDR led to lower expression levels of ATG16L1, lysozyme and LC3 II (figure 4E). Moreover, in a starvation-induced autophagy model, we found that the LC3 II level was significantly lower in VDR−/− MEF incubated in FBS-free E-MEM medium compared with VDR+/− after starvation, although LC3 II was increased by starvation in both VDR−/− and VDR+/− MEF cells (figure 4F). Using lysotracker Red to mark lysosome in vitro,90 we further showed differences in autophagy activity in VDR−/− and VDR+/− MEF cells (figure 4G). Without any treatment, the basal level of lysotracker Red uptake was already lower in VDR−/− MEF compared with VDR+/− cells. After starvation for 2 h to induce autophagy in cells, lysotracker Red uptake was enhanced. However, there was still less autophagy in the cells lacking VDR compared with the VDR+/− MEF cells (figure 4G).
Enhanced VDR protein is associated with elevated autophagy markers within intestinal epithelial cells
Based on our observations, we further determined the possibility for restoration of intestinal epithelial VDR. We hypothesised that the status of VDR changes the expression of autophagy markers. Vitamin D3 is known to increase VDR by blocking ubiquitin/proteasome-mediated degradation.91 To explore the impact of enhanced VDR on autophagy, we investigated the response in the human colonic epithelial cell line INT 407. Vitamin D3 increased LC3 II/I associated with increased VDR in a time-dependent manner. It also increased ATG16L1 associated with elevated level of VDR protein (figure 4H). Activated VDR is known to bind to the VDRE in the promoter of target genes to regulate gene transcription. We further identified the VDRE in the ATG16L1 gene by CHIP assay (figure 4I). Taken together, our data suggested that VDR expression status was able to change autophagy marker ATG16L1, LC3 II and autophagy activity.
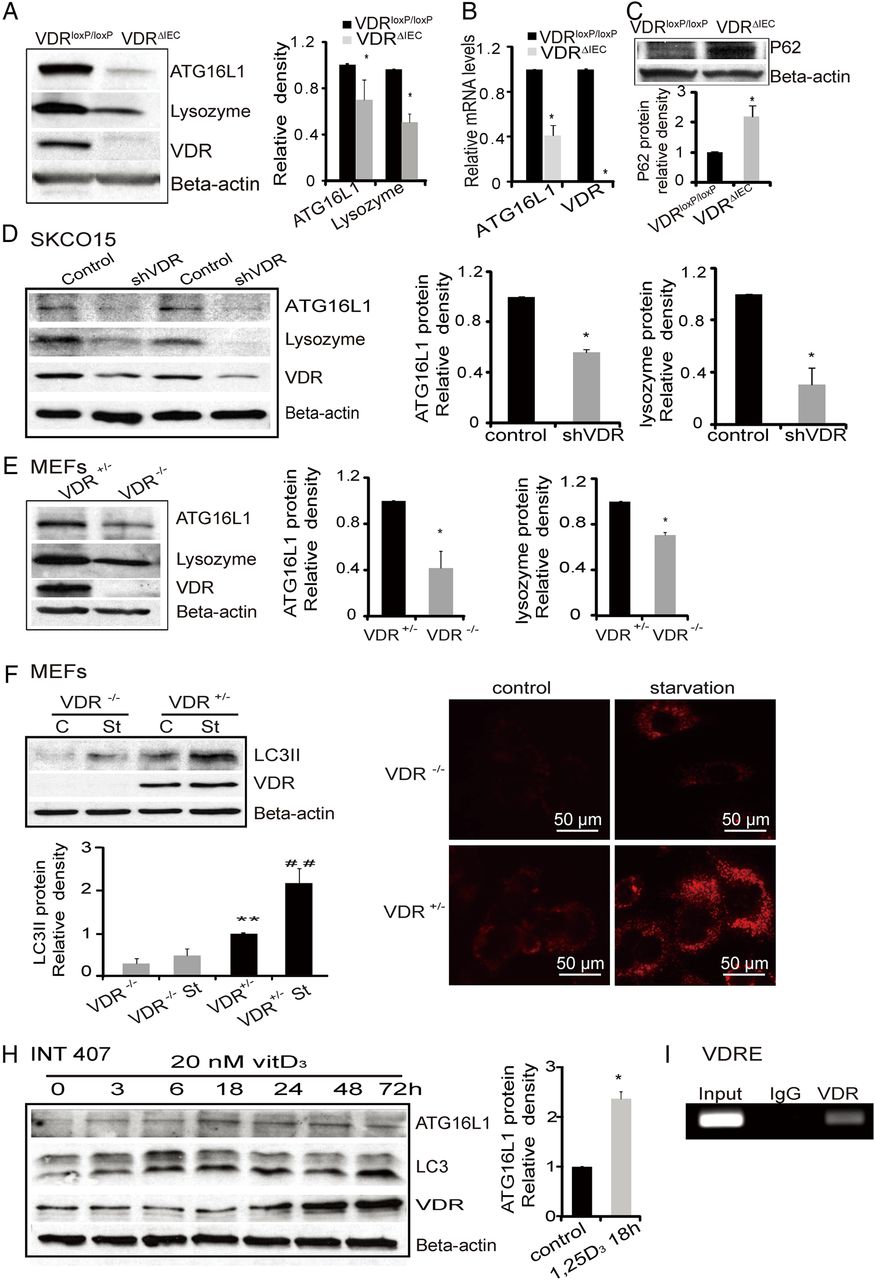
Decreased VDR expression is observed in ulcerative colitis patients and in an experimental colitis IL10−/− model
The VDR is associated with chronic inflammation.8 ,23 Intestinal VDR expression is decreased in patients with ulcerative colitis (UC) as shown in figure 5A, which is consistent with the literature.8 ,23 Moreover, in the inflamed human intestine with low VDR, increased Bacteroides was identified by FISH. Intestinal ATG16L1 staining is also very weak in patients with UC (figure 5C). We found a similar trend of VDR and ATG16L1 expression in an experimental colitis model, where IL10−/− mice spontaneously develop IBD.92 A significant decline in the expression of VDR was found in IL10−/− mice with colitis symptoms compared with mice without colitis symptoms (figure 5D). Moreover, we also found decreased ATG16L1 in intestinal epithelial cells in experimental colitis model (figure 5D).

Vitamin D receptor (VDR) expression in human intestine and colitis models. (A) Intestinal VDR staining is very weak in inflamed colon of patients with ulcerative colitis (UC). (B) Representative fluorescent in situ hybridisation staining for Bacteroides fragilis (red) in tissues from patients with UC. Blue, DAPI. Normal, normal tissue adjacent lesion area; ulcerative colitis, lesion area. (C) Representative intestinal ATG16L1 staining is very weak in inflamed colon of patients with inflammatory bowel disease. (D) Decreased VDR and ATG16L1 in the IL10−/− mice with colitis symptom. Data are presented as the mean±SD from five mice/group.
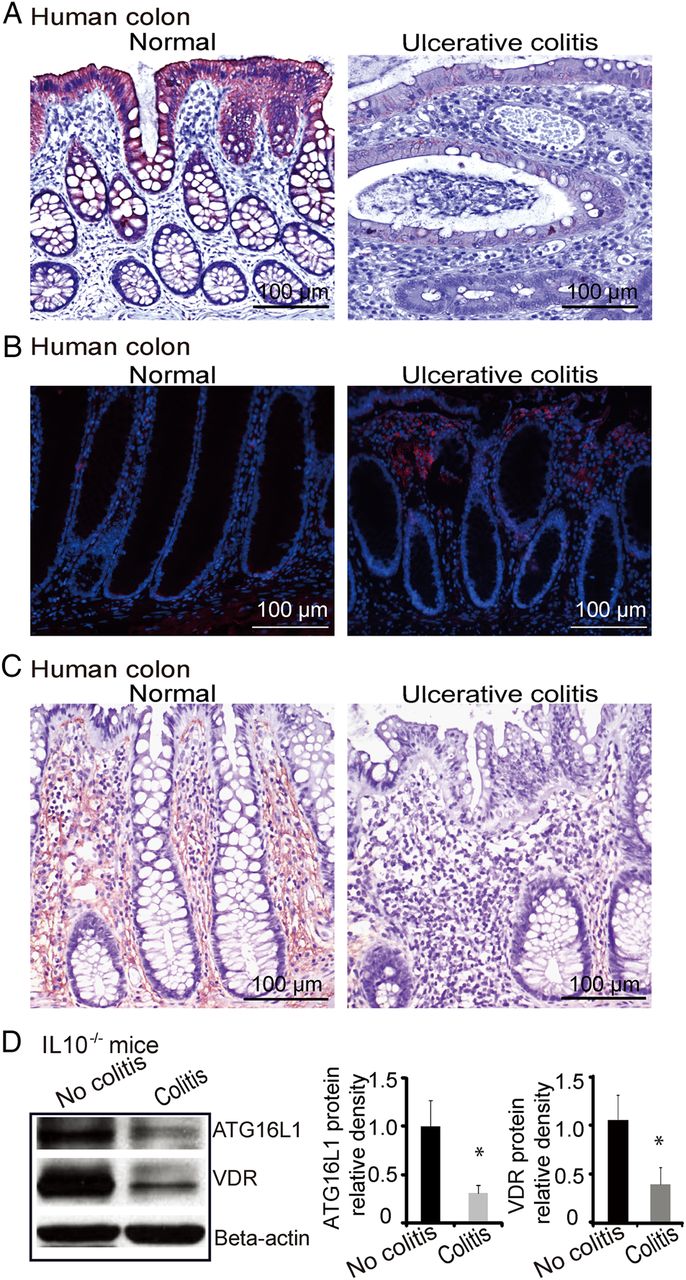
Bacterial product butyrate upregulates VDR in human epithelial cells in vitro
Our microbiome data (figure 1C) indicate that butyrate-producing bacteria (Butyrivibrio) was decreased in the VDRΔIEC mice. Moreover, a recent study indicates that dysbiosis in intestine leads to less butyrate-producing bacteria or less butyrate in intestine.93 We pretreated human intestinal epithelial cells with butyrate and found that it significantly increased VDR expression at the mRNA and protein levels (figure 6A, B). In the human intestinal epithelial cell line HCT116, VDR protein and mRNA were increased by butyrate in a dose-dependent manner (figure 6C, D). We further investigated VDR transcriptional activity following stimulation with butyrate in the human intestinal epithelial cell line HCT116. In cells transfected with the VDR reporter plasmid, butyrate significantly induced VDR transcriptional activity compared with control cells without butyrate stimulation. The relative fold increase was compared with cells transfected with the negative control plasmid (negative) (figure 6E). Additionally, the expression of VDR target genes Cyp27 and cathelicidin increased significantly after butyrate treatment in the human epithelial cells (figure 6F).

Bacterial product butyrate activates vitamin D receptor (VDR) signalling pathway in human intestinal epithelial cells. (A) Butyrate increased VDR protein level in mouse embryonic fibroblast cells. (B) Butyrate increased VDR and lysozyme mRNA levels. (C) Butyrate increased VDR protein level in a dose-dependent manner in HCT116 cells. (D) Butyrate increased VDR mRNA level in a dose-dependent manner in HCT116 cells. (E) Butyrate increased the VDR transcription activity in human epithelial cells. HCT116 cells were transfected with Cignal Vitamin D Reporter (luc) Kit. After transfection for 24 h, cells were treated with 10 mM butyrate for 30 h, and luciferase activity was determined. Firefly luciferase activity was normalised to Renilla luciferase activity, and the activity was expressed as relative luminescence units. (F) Butyrate increased cyp24 and cathelicidin mRNA levels in a dose-dependent manner.
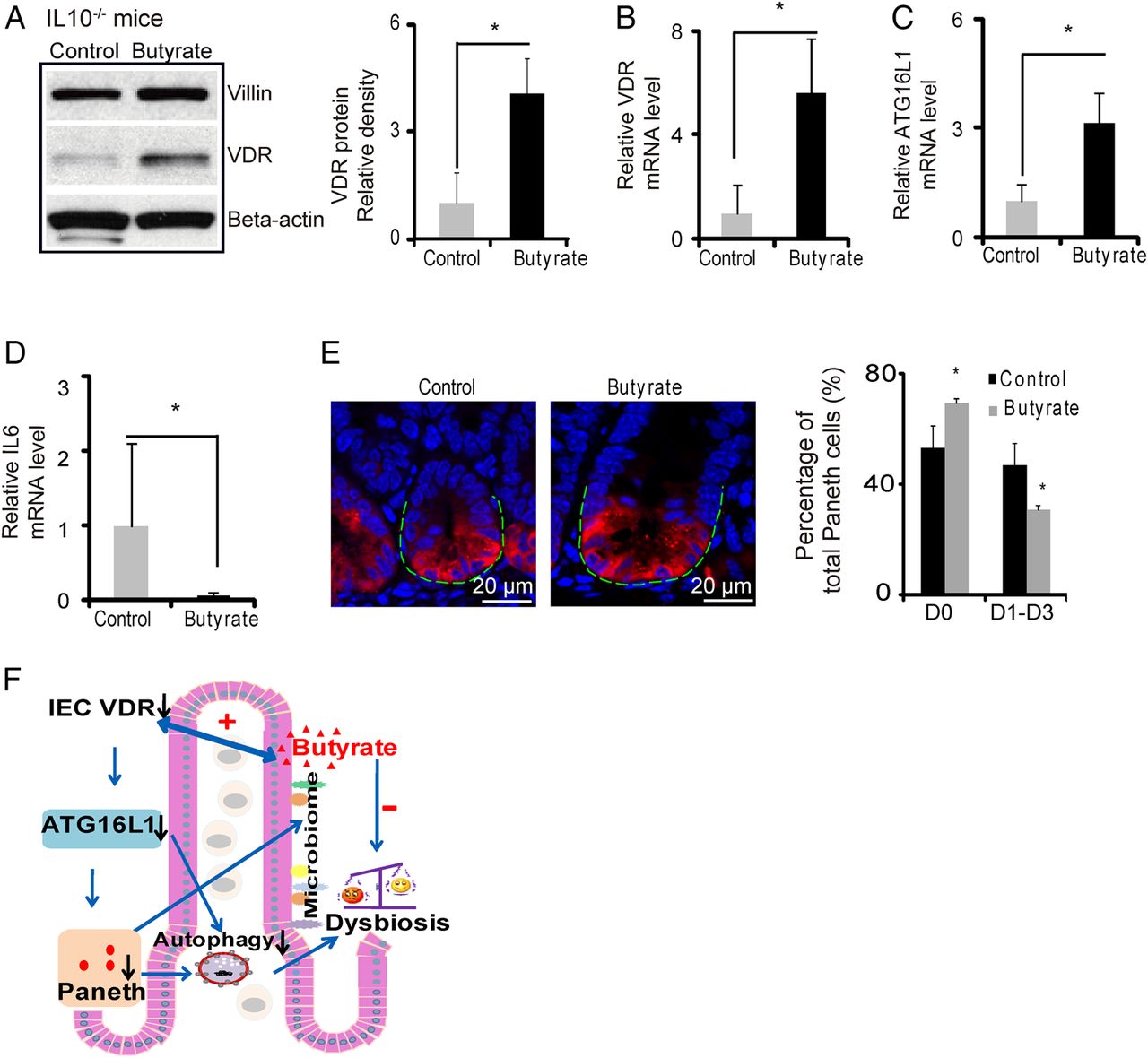
Butyrate treatment increases VDR and suppresses inflammatory cytokine IL-6 in a IL10−/− colitis model
We further used the experimental colitis IL10−/− model to investigate the in vivo physiological relevance of VDR regulation of epithelial function, including responses to bacterial products and anti-inflammatory effects. Butyrate treatment significantly increased VDR at the mRNA and protein levels in the IL-10 KO mice (figure 7A,B). After butyrate treatment, intestinal ATG16L1 was also significantly increased at the mRNA level (figure 7C) and suppression of inflammatory cytokine IL-6 was also observed in the IL-10 KO mice (figure 7D). Meanwhile, butyrate treatment increased the normal Paneth cells ratio (figure 7E). These data suggest that enhancing VDR expression by a bacterial product is able to stimulate autophagy marker ATG16L1, restore number of Paneth cells and suppress inflammation in colitis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Butyrate treatment restores vitamin D receptor (VDR) expression in colitis and inhibits inflammation. (A) Butyrate increased VDR protein in IL10−/− mice. IL10−/− mice were given 2% sodium butyrate in the drinking water for 3 weeks. (B) Butyrate increased VDR mRNA level in IL10−/− mice colon. (C) Butyrate increased ATG16L1 mRNA levels in IL10−/− mice. (D) Butyrate decreased IL6 mRNA level (n=4, *p<0.05). (E) Representative images of indirect immunofluorescence of sections stained for lysozyme (red) in ileal crypts of IL10−/− mice with/without butyrate treatment. Percentage of Paneth cells displayed normal and abnormal (D1 to D3) patterns of lysozyme expression (n=4/group, *p<0.05). (F) A working model of VDR in intestinal and microbiome homeostasis.
Discussion
The positive association of vitamin D deficiency and VDR polymorphisms with IBD suggests that vitamin D metabolism and VDR likely plays a vital regulatory role in this disease.94 ,95 Our study and others indicate that VDR deletions exaggerate colitis through activation the NF-κB pathway.25 ,26 However, the exact role of intestinal VDR in IBD and inflammation remains elusive. In the current study, we established a VDRΔIEC mouse model, with selective KO of VDR expression in intestinal epithelial cells, to investigate the role of VDR on intestinal inflammation, autophagy, antimicrobial peptide expression and microbiota composition. This study shows that VDR acts as a master regulator of intestinal homeostasis and establishes a unifying link between VDR, autophagy, the intestinal microbiota and innate immunity, all factors that have been implicated in the pathogenesis of IBD.
Many studies report a link between autophagy and IBD,96 ,97 and in a study of the intracellular bacterium M. tuberculosis, 1,25-dihydroxyvitamin D3 was shown to induce autophagy in human monocytes.61 Our previously published work with Salmonella typhimurium, another intracellular pathogen, indicated that VDR-null mutant mice have worse outcomes with Salmonella-induced infection than WT controls.65 Since autophagy is important in host defence and to help limit systemic dissemination of Salmonella, we sought to investigate the specific impact of an intestinal VDR deletion on autophagy. Significantly lower amounts of ATG16L1 were found in VDRΔIEC compared with the VDRloxP/loxP controls at both the protein and the transcriptional levels, while leaving the other regulators of autophagy intact (eg, mTOR, phospho-p70 S6 kinase, Beclin-1, data not shown). ATG16L1 is one of the ‘core ATG proteins’ required to form the ATG5-ATG12-ATG16L1 complex to promote elongation and closure of the autophagosome.98 Lack of ATG16L1 led to autophagy deficiency in mouse cells.58 ,59 Lysozyme, one of the proteases in lysosomes, is associated with autophagy maturation.99 ,100 Our data showed VDR KO decreased lysozyme in VDR−/− MEF cells and ileal epithelial cells. This data further indicated that loss of VDR protein may result in a defect of autophagy maturation in cells. Others have similarly reported lack of lysozyme staining in the mucus of ATG16L1 mutant mice.13
Lysozyme is also a component and marker of the Paneth cell secretory granule. Paneth cells are specialised epithelial cells primarily located in the small intestine. Paneth cells secrete granules containing antimicrobial peptides including lysozyme.101–103 Our data showed that the total amount and normal expression pattern of Paneth cell decreased in VDR−/− mouse ileum. Defective lysozyme and Paneth cell in VDRΔIEC intestine indicates an abnormality of Paneth cell secretion, particularly in the granule exocytosis pathway. VDR−/− ileum contained an increased proportion of Paneth cells with disorganised or diminished granules or exhibiting diffuse cytoplasmic lysozyme staining. The total amount of Paneth cells also decreased in VDR−/− mice. Interestingly, an ATG16L1 mutation was shown to confer Paneth cell defects in Norovirus-infected mice as well as patients with CD.59 Hence, loss of normal ATG16L1 gene function may result in aberrant Paneth cell function, suggesting that Paneth cell dysfunction may play an important role in IBD.59 Another gene linked to IBD, XBP1 has also been implicated in severe Paneth cell and goblet cell dysfunction.104 These studies are consistent with our findings of aberrant Paneth cell function, suggesting that the loss of the ability to secrete antimicrobial peptides may be an important factor in the development of intestinal inflammation and IBD.
The intestinal Paneth cells are known to secrete antimicrobial peptides and shape the composition of the microbiome.87 ,88 Paneth cells play a key role in establishing and maintaining the intestinal microbiota, and the ability of Paneth cell alpha defensins to remodel the gut microbiota communities has been well described.105 VDRΔIEC mice were found to have increased bacterial loads and a shift of species (increased E. coli and Bacteroides and decreased butyrate-producing bacteria) and developed a more severe DSS-induced colitis compared with the VDRloxP/loxP mice, suggesting that microbial dysbiosis in VDRΔIEC mice may sensitise the colonic mucosa to chemical injury induced by DSS. It is possible that a defect in this first-line mucosal clearance of microbes may ultimately contribute to excessive chronic inflammatory responses. In the IL10−/− experimental colitis model, we were able to increase intestinal VDR expression by administration of the bacterial metabolite, butyrate and decrease IL-6 inflammatory cytokine expression, consistent with what has been described by others.106
Our data showed dysbiosis, including decreased abundance of Butyrivibrio, in VDRΔIEC mice increase risk for colitis. Dysbiotic microbial ecology is known to contribute to the pathogenesis of IBD and could be corrected by faecal transplantation, based on a recent study using Nod2-deficient mice.69 Our co-housing data indicate that the absence of intestinal epithelial VDR confers a transmissible risk for DSS-induced colitis. Therefore, for the VDR-deficient mice, there are several potential strategies to correct colitis by (1) treating mice with butyrate, a bacterial natural product; (2) enhancing intestinal VDR expression; and (3) faecal transplantation. We will explore faecal transplantation in a future study.
In skin, cathelicidin is a direct target of VDR and is upregulated by vitamin D.107 Vitamin D is thought to be an important regulator for antimicrobial peptides found in skin, but our study indicates that VDR may be similarly important for AMP expression in the intestine. The regulatory networks that control AMP expression still need to be fully defined, but our findings may have important implications not only for understanding the mechanisms of vitamin D and microbiota remodelling in the gut, but also for other epithelial cell-based autoimmune diseases such as psoriasis 108 where dysbiotic microbial communities and aberrant immune responses may similarly play a role in pathogenesis.
In conclusion, our data indicate that intestinal VDR plays a fundamental role in intestinal homeostasis through its effects on autophagy, on Paneth cells and on the intestinal microbiota itself. There are mutual interactions between vitamin D, VDR and the microbiome that still remain to be fully understood. However, our study suggests that intestinal VDR may represent a master regulator of inflammation by regulating autophagy and the production of antimicrobial peptides that, in turn, are responsible for remodelling the bacterial communities that comprise the intestinal microbiota. All of these factors (inflammation, autophagy, microbiota composition) have been implicated in the pathogenesis of IBD; we propose that all of these factors may in fact be regulated under the control of one master regulatory pathway, through VDR (figure 7F). Although the complex regulatory network that controls each of these aspects still needs to be fully elucidated, our findings are important not only for a better understanding IBD but may also have applicability for other autoimmune diseases (such as those of the skin, lung), where the host is in contact with bacteria, and aberrant immune responses and microbiota dysbiosis are implicated. Insights gained from understanding how the VDR pathway is integrally involved in regulating autophagy and changing microbiome diversity may serve as a paradigm for understanding the nature of host defence signals in inflammation.
Acknowledgments
We thank Liesbet Lieben for technical assistance with VDRΔIEC mice. This work was supported by the NIDDK (KO1 DK075386 and 1R03DK089010–01), the American Cancer Society (RSG-09-075-01-MBC) and Swim Across America Cancer Research Award to JS. NIDDK DK42086 (DDRCC), DK097268 and DK47722 to EBC.
References
Footnotes
Contributors Contributions to the conception or design of the work; or the acquisition, analysis or interpretation of data for the work: SW, YZ, RL, YX, DZ, DC and JS. Providing critical experimental material, reagents, drafting the work or revising it critically for important intellectual content: SW, YZ, RL, YX, DZ, EOP, EC, DC, EBC, GC and JS. Final approval of the version to be published: SW, YZ, RL, YX, DZ, EOP, EC, DC, EBC, GC and JS. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved: SW, YZ, RL, YX, DZ, EOP, EC, DC, EBC, GC and JS.
Competing interests None.
Ethics approval University of Rochester Ethics Committee (RSRB00037178).
Provenance and peer review Not commissioned; externally peer reviewed.