Article Text

Abstract
Background and objective While pro-inflammatory monocyte trafficking to the intestine has been partially characterised, the molecules required for migration of tolerogenic mononuclear phagocytes (dendritic cells (DC) and macrophages) are unknown. We hypothesised that the gut-homing receptor integrin α4β7 is required for this process.
Methods We used a T cell-mediated colitis model to study the role of α4β7 in the innate immune compartment. We then performed competitive bone marrow (BM) reconstitution experiments to assess the requirement of α4β7 in the generation of intestinal retinoic acid (RA)-producing CD11chi DC (ALDE+DC) and CD64 macrophages. Using mixed BM chimeras we also asked whether α4β7 is required to give rise to tolerogenic mononuclear phagocytes.
Results Lack of β7 integrins in the innate immune compartment (β7−/−RAG2−/− mice) markedly accelerated T cell-mediated colitis, which was correlated with lower numbers and frequencies of ALDE+DC in mesenteric lymph nodes. Consistent with a role of α4β7 in the generation of intestinal mononuclear phagocytes, BM cells from β7−/− mice poorly reconstituted small intestine ALDE+DC and Mφ when compared to their wild type counterparts. In addition, mice lacking β7 integrins in the CD11chi compartment showed decreased ability to induce Foxp3+ TREG and IL-10-producing T cells.
Conclusions Mice lacking β7 integrins in the innate immune compartment are more susceptible to intestinal inflammation, which is correlated with a requirement of β7 integrins to reconstitute gut mononuclear phagocytes with tolerogenic potential.
- DENDRITIC CELLS
- ADHESION MOLECULES
- TOLERANCE
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
-
Integrin α4β7 is required for T and B cell migration to the gut mucosa
-
Retinoic acid (RA) regulates the development of gut-dendritic cells (DC) precursors
-
RA-producing DC (ALDE+DC) induce gut-tropic lymphocytes and promote TREG differentiation
What are the new findings?
-
Lack of α4β7 in the innate immune compartment accelerates T cell-mediated colitis, which is correlated with a decrease in ALDE+DC
-
α4β7 is required for reconstitution of intestinal mononuclear phagocytes, in particular ALDE+DC
-
A subset of bone marrow (BM) cell progenitors expresses α4β7 and can give rise to ALDE+DC
-
DC expressing α4β7 are required to induce TREG and IL-10-producing T cells in vivo
How might it impact on clinical practice in the foreseeable future?
-
We show that BM progenitors require the integrin α4β7 to give rise to intestinal ALDE+DC with tolerogenic potential. This study might contribute to the development of DC-based therapies to boost immune tolerance during intestinal inflammation.
Introduction
Mononuclear phagocytes, including dendritic cells (DC) and macrophages (Mφ), are enriched in the intestinal mucosa and contribute to the balance between pro-inflammatory and tolerogenic immune responses in this compartment. Changes in DC and Mφ composition or functional properties might result in immune deregulation, such as during inflammatory bowel diseases.1 ,2 Two main CD11c-expressing mononuclear phagocytic subsets have been described in the intestinal lamina propria (LP). The CX3CR1hi cells are considered to be Mφ, whereas CD103 cells are DC.2–5 In addition, a subset of CD103 DC have the ability to synthesise all-trans retinoic acid (RA), which is sufficient to induce α4β7+CCR9+ gut-homing T and B cells, and in combination with TGF-β enhances TREG differentiation.6 Furthermore, recent data indicate that RA-dependent induction of gut-tropic T cells is critically required to induce oral immune tolerance.7 ,8
The chemokine receptor CCR2 is involved in the recruitment of monocytes to the healthy and inflamed colon, which then differentiate to Mφ and migratory antigen presenting cells with DC morphology.9 ,10 Ablation of CCR2+ monocytes is protective in dextran sulfate sodium-induced colitis,10 suggesting that CCR2 plays a major role in the generation of pro-inflammatory mononuclear phagocytes in the gut. However, the migratory requirements of intestinal tolerogenic mononuclear phagocytes remain unknown.
Here, using a T cell-mediated colitis model we show that β7 deficiency in the innate immune compartment results in accelerated induction of intestinal inflammation, which is associated with a reduction of RA-synthesising DC in mesenteric lymph nodes (MLN). Moreover, bone marrow (BM) progenitors from mice lacking the β7 integrin chain (required for α4β7 and CD103/αEβ7 integrins) are impaired in reconstituting the small intestine mononuclear phagocyte compartment, including RA-producing DC. Importantly, we show that β7 integrins are required to give rise to mononuclear phagocytes with the capacity to generate antigen-specific Foxp3+ TREG and IL-10 producing cells on oral antigen administration.
Materials and methods
Mice
Wild type (wt) CD45.1/CD45.2, CD11c-DTR, RAG2−/−, CXCR3−/− and β7−/− mice were purchased from Jackson Laboratories (Bar Harbor, Maine, USA). CCR9−/− mice were provided by Dr Paul Love.8 Flt3−/− mice were provided by Dr Christoph Schaniel (Mount Sinai School of Medicine, New York, USA).11 All strains were on the C57BL/6 background and used at 8–13 weeks old. Mice were maintained in SPF/VAF conditions and used in accordance with the guidelines from the Subcommittee on Research Animal Care at Massachusetts General Hospital and Harvard Medical School.
Competitive BM reconstitution experiments
Whole or fractionated BM from wt CD45.1 mice were mixed with the congenic counterpart (CD45.2) in a 1:1 or 1:2 ratio, then transferred intravenously into CD45.1/2 recipients that were previously irradiated (unless otherwise indicated). Mice were sacrificed after 8 weeks and single cell suspensions were generated from spleen, BM, MLN, peripheral lymph nodes, Peyer's patches (PP) or colon and small intestine LP. LP cells were isolated after carefully removing the PP and the caecal patch, as described.12 Cell samples were stained as indicated and analysed on a fluorescence-activated cell sorter (FACS) LSRII (BD Biosciences). The reconstitution index (RI) was calculated as: RI=[wt DC]tissue/[KO DC]tissue: [wt DC]spleen/[KO DC]spleen.
Isolation of BM cell populations
BM cells were harvested from the femora and tibiae of wild type CD45.1 or β7−/− mice and enriched for the lineage (Lin: CD11c, PDCA-1, F4/80, CD3, NK1.1, B220, CD19, and TER119) negative fraction by immunomagnetic technique using anti-biotin magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany). The cells were then immunostained with anti-Ly6C and Lin and then purified by high-speed sorting with a FACS Aria (BD Biosciences). Pure populations were injected intravenously into congenic CD45.1/2 diphtheria toxin (DT)-treated CD11c-DTR BM chimeras.
Reconstitution of DC-depleted mice
For DC depletion, CD11c-DTR BM chimeras were treated intraperitoneally with 160 ng/mouse DT 1 day before LinnegBM cell transfer and then every other day for 7 days. One million sorted LinnegLy6Clow or LinnegLy6Chi cells in 200 μL phosphate-buffered saline (PBS) were injected intravenously.
Statistical analysis
Data are presented as mean±SEM and were analysed using GraphPad Prism Software V.5.0d. Data were analysed using t tests for comparing two groups or one-way analysis of variance (ANOVA) with Dunnett's post-hoc test when comparing more than two groups.
Results
Characterisation of ALDE+DC during intestinal inflammation
Owing to their ability to induce Foxp3+TREG through mechanisms involving TGF-β and RA, intestinal CD103 DC from MLNs (MLN-DC) have been considered to have tolerogenic potential.3 Interestingly, we noticed that a fraction of CD103 MLN-DC are negative for Aldefluor (ALDE) staining (figure 1A), suggesting that these DC lack retinal dehydrogenase (RALDH) activity and cannot produce RA. To functionally explore this possibility, we sorted CD103+ALDE+ and CD103+ALDEneg MLN-DC and tested their ability to induce gut-homing receptors on CD8 T cells, which requires RA.13 Consistent with our hypothesis, only CD103+ALDE+ MLN-DC induced CCR9 or α4β7 on CD8 T cells, whereas CD103+ALDEneg MLN-DC did not induce gut-homing receptors, a deficit that was rescued by the addition of exogenous RA (figure 1A). These results are in agreement with a report showing that the capacity of ALDE+DC to potentiate TREG induction was not correlated to their CD103 expression.14
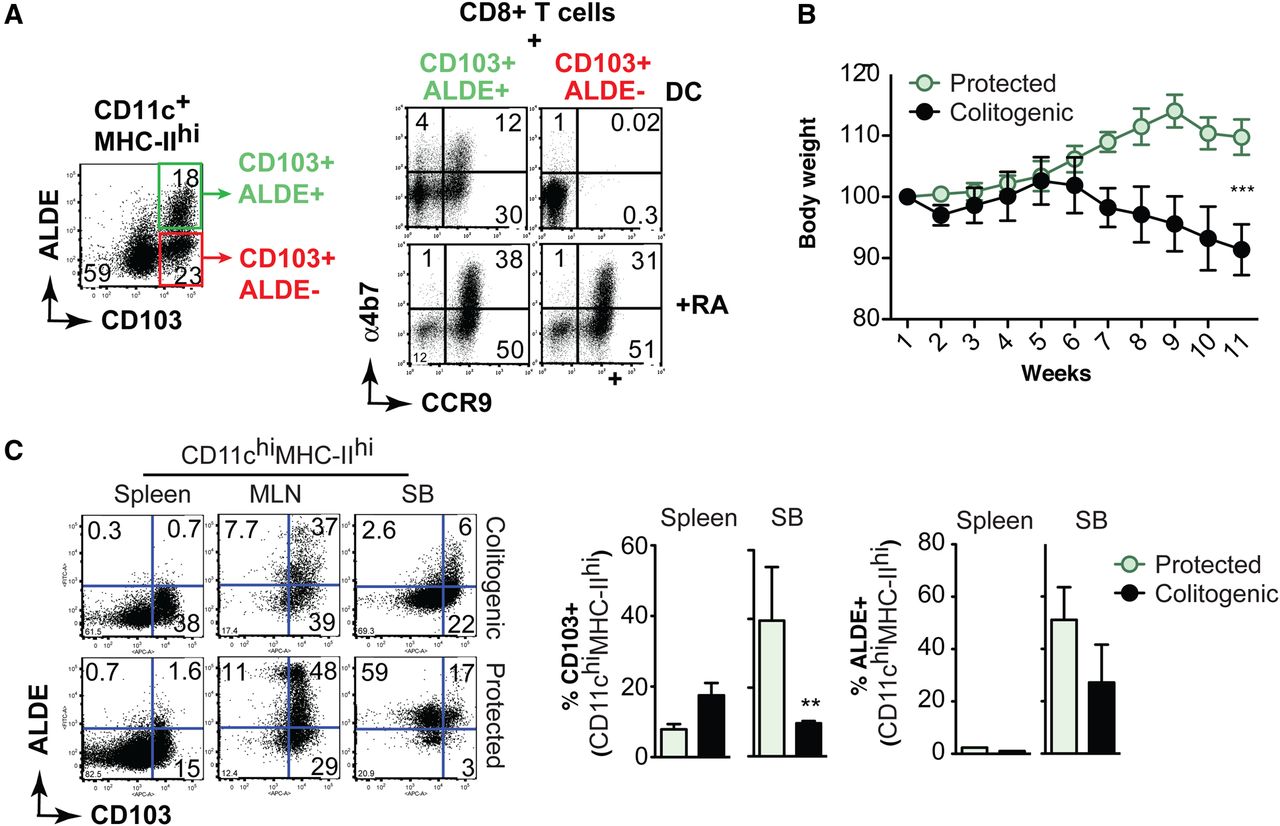
Decreased frequency of ALDE+ dendritic cells (DC) during intestinal inflammation. (A) Flow cytometry analysis of CD11c+MHC-IIhi cells from mesenteric lymph nodes (MLN) stained for CD103 and Aldefluor (ALDE). ALDE+ (CD103+ALDE+) and ALDEneg (CD103+ALDE−) MLN-DC were sorted (left panels) and incubated with CFSE-labelled OT-I CD8 T cells in the presence of ovalbumin peptide±all-trans retinoic acid (RA). After 5 days of culture T cells were analysed for the expression of CCR9 and α4β7 (right panels). Data are representative of three experiments. (B and C) RAG2−/− mice received wild type CD4+CD25negCD45RBhi naïve T cells alone (colitogenic) or combined with CD4+CD25+CD45RBlow TREG (protected). (B) Body weight loss (mean±SEM; 6 mice/group). (C) Staining for CD103 and ALDE on CD11chiMHC-IIhi small intestine lamina propria (SI-LP) cells (mean±SEM; 3 mice/group). **p<0.01, ***p<0.001, unpaired t test.
Then, we assessed whether ALDE staining correlates with CD103 expression in DC from mice undergoing intestinal inflammation. Recombination activating gene 2 deficient (RAG−/−) mice were adoptively transferred with CD4+CD25−CD45RBhi naïve T cells alone (colitogenic) or combined with CD4+CD25+CD45RBlow TREG cells (protected), as described previously15 ,16 (figure 1B). We observed that both CD103+DC and ALDE+DC were decreased in the small intestine propria (SI-LP), but not in the spleen, of colitogenic mice when compared to protected animals (figure 1C). Interestingly, we observed dissociation between CD103 and ALDE staining in SI-LP DC from protected mice (figure 1C). Thus, our data indicate that CD103 and ALDE staining do not always correlate, and that ALDE staining might be a better strategy to functionally label RA-producing DC with tolerogenic potential.
Lack of β7 integrins in the innate immune compartment accelerates T cell-mediated colitis
The integrin α4β7 has been largely characterised as a gut-homing receptor for adaptive immune cells, and blockade of α4β7/MAdCAM-1 decreased T cell infiltration and intestinal inflammation in a T cell-mediated colitis model.17 We asked whether lack of α4β7 in the innate immune compartment might also affect intestinal inflammation in a T cell-mediated colitis model. We crossed β7−/− mice with RAG−/− to generate β7−/−RAG−/− mice. RAG−/− or β7−/−RAG−/− mice were adoptively transferred with wt CD4+CD25−CD45RBhi T cells and then assessed for colitis. RAG−/− mice started to lose body weight about 30 days after T cell transfer (see online supplementary figure S1). Unexpectedly, β7−/−RAG−/− mice started to lose weight as fast as 7–10 days post T cell transfer (figure 2A). By day 14, ±30% of β7−/−RAG−/− mice exhibited rectal prolapse and bleeding and ±80% showed marked body weight loss. In addition, β7−/−RAG−/− mice exhibited higher colitis scores and inflammation compared to RAG−/− mice (figure 2B,C). Previous reports showed that MLN-DC from colitic mice (day 14) have reduced capacity to produce all-trans RA or TGF-β, and were impaired for generating Foxp3+ TREG.18 Analogously, using the ALDE assay to assess RA-producing capacity,12 ,19 we showed that colitic β7−/−RAG−/− mice have lower frequencies and numbers of ALDE+MLN-DC when compared to RAG−/− mice (figure 2D–F). Of note, analysis of ALDE+DC and Mφ before T cell transfer (day 0) showed no difference between β7−/−RAG−/− and RAG−/− mice in the SI-LP (see online supplementary figure S2), whereas we found a small decrease in ALDE+MLN-DC in β7−/−RAG−/− mice (figure 2E,F). Thus, whereas α4β7 is required in T cells for infiltrating the gut and promoting inflammation,17 this integrin appears to be required in the innate immune compartment to fulfil a tolerogenic role in the context of T cell-mediated colitis, which correlates with the presence of ALDE+DC.

Lack of β7 integrins in innate immune cells predisposes to T cell-mediated colitis. RAG−/− or β7−/−RAG−/− mice were adoptively transferred with 0.4×106 wt CD4+CD25−CD45RBhi naïve T cells and then assessed for colitis development. (A) Mean body weight (n=6 mice per group). (B) Clinical scores (0–6) at day 14 post T cell transfer. (C) Colitis scores (0–9) and representative photomicrograph of proximal colon sections at day 14 post T cell transfer. (D–F) Aldefluor (ALDE) staining of mesenteric lymph nodes (MLN)-dendritic cells from RAG−/− or β7−/−RAG−/− at day 14 post T cell transfer (n=3). Mean±SEM, *p<0.05, **p<0.001. (A) One-way ANOVA. (B) Unpaired t test.
BM cells require β7 integrins to reconstitute RA-synthesising CD11c mononuclear phagocytes in the small intestine
To directly assess whether β7 integrins are required for ALDE+DC intestinal reconstitution, we compared BM cells from wt mice versus cells from mice deficient in different adhesion and/or chemokine receptors in terms of their capacity to reconstitute gut-associated mononuclear phagocytes. We assessed reconstitution of CD11c mononuclear phagocytes (see online supplementary figure S3) in lethally irradiated mice 8 weeks post BM transfer. Although CCR9−/− and CXCR3−/− BM cells reconstituted DC at a similar level than wt cells (RI close to 1), BM cells from β7−/− mice were impaired in reconstituting CD11c mononuclear phagocytes in MLN (figure 3A,B). Of note, the role of β7 integrins in mononuclear phagocyte and plasmacytoid DC (pDC, CD11cintB220+) reconstitution was much more pronounced in PP and SI-LP compared to MLN, whereas CCR9 did not play a significant role in pDC or DC reconstitution in these tissues (figure 3C, see online supplementary figure S4, and data not shown). Unexpectedly, β7 integrins were not required to give rise to colon CD11c mononuclear phagocytes (figure 3C). CD11c include both DC and Mφ.20 Thus, we sought to investigate whether β7 integrins are required to reconstitute DC and/or Mφ. Although DC can be distinguished from Mφ by their expression of CD103, we were not able to use CD103 as a DC marker in β7−/− cells (β7 integrin chain is required for CD103/αEβ7 cell surface expression). However, since only CD103 DC express RALDH enzymes21 we considered ALDE+ cells as DC,12 ,19 whereas CD11c+ALDEneg cells could be either DC or Mφ. To further discriminate between ALDEneg Mφ and DC we used CD64 (FcγRI), a recently described marker for gut-associated Mφ.22 ,23 Wt (CD45.1) and β7−/− (CD45.2) BM were transferred in a 1:2 ratio (to favour visualisation of KO cells) into lethally irradiated recipients (CD45.1+/−) and after 8 weeks we analysed the RI. We did not detect CD11c+ALDE+DC or CD64+ Mφ in the spleen (data not shown), whereas MLN and SI-LP revealed the presence of CD11c+MHC-IIhiALDE+ and ALDEnegDC, as well as ALDEneg CD64 Mφ (figure 3D,E and see online supplementary figure S3). When analysing each population of mononuclear phagocytes, our data showed that β7 integrins are mostly required to give rise to ALDE+DC and to a lesser extent ALDEnegDC, while CD64 Mφ were not significantly compromised (figure 3D,E).

Bone marrow (BM) cells require β7 integrins to reconstitute CD11c mononuclear phagocytes in the small intestine lamina propria (SI-LP). (A and B) BM cells from wt (CD45.1/2, expressing both CD45.1 and CD45.2) and from β7−/−, CCR9−/−, β7−/−CCR9−/−, CXCR3−/− or Flt3−/− (KO, expressing CD45.2) mice were transferred in a 1:1 ratio into lethally irradiated recipients (CD45.1). Eight weeks later cells were isolated from the indicated tissues and analysed for the proportion of wt and KO CD11c cells. Data are presented as reconstitution index (RI), defined as the ratio between wt and KO MHC-II+CD11c+ cells in each organ divided by the same ratio in the spleen. (A and B) RI comparing wt versus KO in mesenteric lymph nodes (MLN) (n=4–8). (C) RI comparing wt versus β7−/− BM in spleen, BM, peripheral lymph nodes (PLN), MLN, Peyer's patches (PP), SI-LP, and colon (n=4–8). Three independent experiments (n=2–3 mice/experiment). (D and E) BM cells from wt (CD45.1) and from β7−/− (CD45.2) mice were transferred in a 1:2 ratio (wt:β7−/−) into lethally irradiated recipients (CD45.1/2). Eight weeks later cells were isolated from spleen, MLN and BM (D), SI-LP and colon (E), and analysed for the proportion of wt and β7−/− cells. CD11c cells were subdivided into Mφ (CD64) and dendritic cells (DC) (CD64neg). DC were further divided into retinoic acid (RA)-producing (ALDE+) or non-RA-producing (ALDEneg) DC. Graph colours indicate blood cells (white), CD11cnegMHC-IIneg cells (black), Mφ (red), non-RA-producing DC (blue) and RA-producing DC (green) (n=3). Two independent experiments (n=1–2 mice/experiment). Mean±SEM, *p<0.05, **p<0.01, ***p<0.005 (one-sample t test vs hypothetical RI=1.0). Red dashed lined indicate RI=1 (equivalent migration between wt and β7−/− cells).
LinnegLy6Clow BM cells express gut-homing receptors and give rise to RA-producing DC
Although Mφ and DC can be found in the BM,24 BM precursors, rather than terminally differentiated Mφ/DC, have been shown to reconstitute intestinal LP DC.11 ,25 In fact, we did not detect high expression of α4β7 or ALDE+ cells within the BM CD11c population (figure 4A and data not shown). Further analysis of the CD11cneg population revealed that lineage (Lin) negative (Linneg)Ly6Clow BM cells contained distinct populations expressing α4β7 and/or CCR9 (figure 4B). Moreover, we found that α4β7+ is also expressed in a small population of pre-DC (figure 4C). Although it has been shown that LinnegLy6Clow BM cells differentiate to CD103+DC within the LP,11 ,25 whether they also differentiate into RA-producing DC has not been investigated. To address this question, we adoptively transferred sorted LinnegLy6Clow or LinnegLy6Chi BM cells into DC-depleted mice. For this purpose, we previously reconstituted lethally irradiated mice with BM from CD11c-DTR mice, hence making CD11c cells sensitive to DT.26 After 7 days of DT treatment we observed efficient depletion of SI-LP and PP CD11c cells (figure 4D). As expected, LinnegLy6Clow cells, but not LinnegLy6Chi monocytes, gave rise to ALDE+DC (figure 4D). We then sought to investigate whether LinnegLy6Clow cells require β7 integrins to give rise to ALDE+ MLN-DC. We mixed wt (CD45.1) and β7−/− (CD45.2) BM cells in a 1:1 ratio, sorted LinnegLy6Clow and transferred into chimeric CD11c-DTR recipient mice (CD45.1/2, expressing both CD45.1 and CD45.2) that were treated with DT every other day for 7 days. Wt LinnegLy6Clow cells showed a significant advantage in reconstituting ALDE+ and ALDEneg MLN-DC when compared to their β7−/− counterparts, although this difference tended to be more pronounced for ALDE+DC (figure 4E). Of note, wt and β7−/− cells gave rise to similar numbers of spleen DC in vivo (figure 3C,D) and we generated the same frequencies of DC in vitro when using either wt or β7−/− BM cells (see online supplementary figure S5A,B). Furthermore, on feeding RA12 we induced comparable levels of RA-producing extra-intestinal DC in wt and β7−/− mice (see online supplementary figure S5C,D). These data indicate that β7 integrins are not intrinsically required for DC differentiation or for their education to produce RA.

LinnegLy6Clow bone marrow (BM) cells express gut-homing receptors and give rise to gut-associated dendritic cells (DC). (A and B) Expression of α4β7 and CCR9 in BM cell subsets. CCR9−/− and β7−/− cells were used as negative staining controls. (A) DC (CD11c+B220neg), pDC (CD11c+B220int) and CD11cneg cells. (B) Lineage (TER119, CD19, CD11c, PDCA-1, F4/80, NK1.1, CD3) negative (Linneg) and Ly6Chi or Ly6Clow cells. (C) Expression of α4β7 in pre-DC (LinnegCD11c+CD135+MHC-IIneg) (n=3). (D) Sorted LinnegLy6Chi or LinnegLy6Clow cells (CD45.1) were adoptively transferred into CD11c-DTR mice (CD45.1/2) that were treated or not with diphtheria toxin (DT) every other day for 7 days. After that the mice were analysed for the presence of CD45.1+CD11c+ cells and RALDH activity (n=4). Mean±SEM, *p<0.05 (ANOVA vs the second column). (E) BM cells from wt (CD45.1) and β7−/− (CD45.2) mice were sorted for LinnegLy6Clow cells and then transferred into CD11c-DTR mice (CD45.1/2) that were treated or not with DT every other day for 7 days. Mesenteric lymph node (MLN) cells were then stained for CD11c, CD45.1, CD45.2, and analysed for RALDH activity (Aldefluor (ALDE)). Cells gated in ALDE+ or ALDEneg CD11c DC were analysed for their ratio of wt and β7−/− cells and normalised using the spleen ratio (n=5–6). Mean±SEM, *p<0.05, ***p<0.005 (ANOVA vs spleen DC).
RA is required for the generation of α4β7+ LinnegLy6Clow BM cells
RA is required to confer gut-associated DC with RA-producing capacity.12 ,27 ,28 Thus, to explore whether RA is also involved in the generation of LinnegLy6Clow BM progenitors expressing α4β7, mice were maintained on a vitamin A-deficient (VAD) diet, hence eliminating the substrate for RA biosynthesis. While VAD mice exhibited only a slight decrease in the proportions of common DC precursors (CDP) and macrophage DC precursors (MDP) (figure 5A), the percentages of α4β7+ (and α4β7+CCR9+) LinnegLy6Clow BM cells were markedly reduced when compared to mice on a control vitamin A-sufficient diet (figure 5B and see online supplementary figure S6). Among LinnegLy6Clow BM cells, CDP and MDP also showed a reduction in α4β7 expression in VAD mice (figure 5C). Of note, while VAD mice exhibited only a slight reduction in the proportion of CD103 DC in MLN (figure 5D), they showed a marked decrease in ALDE+DC in MLN (figure 5E), suggesting that α4β7+ LinnegLy6Clow cells are especially required to give rise to RA-producing DC. Overall, our results suggest that RA induces α4β7 in a subset of LinnegLy6Clow DC/Mφ precursors, conferring these cells with gut-homing capacity.

Role of retinoic acid in the generation of α4β7+ LinnegLy6Clow bone marrow (BM) cells. (A–C) BM cells from control or vitamin A-depleted (VAD) mice were gated on LinnegLy6Clow cells and stained for (A) c-kit and CD135, or (B) α4β7 and CCR9 (n=5). (C) BM analysis of α4β7 expression in MDP (CD135+c-kit+), CDP (CD135+c-kit−) and negative fraction (CD135−c-kit−) previously gated in LinnegLy6Clow (n=3). (D) CD103 expression on CD11c dendritic cells (DC) in mesenteric lymph nodes (MLN) from control or VAD mice (n=5). (E) MLN cells from control or VAD mice were gated on CD11c cells and analysed for CD103 expression and RALDH activity. FACS plots are representative of three independent experiments. Mean±SEM, *p<0.05, ***p<0.001 (unpaired t test).
Although the generation of α4β7+LinnegLy6Clow BM cells requires RA, the source of RA in the BM remains to be determined. Human haematopoietic stem cells (HSC) exhibit high RALDH activity,29 suggesting that HSC might be a potential source of RA in the BM. However, we did not observe high ALDE staining in LinnegLy6Clow cells from wt mice (see online supplementary figure S7).
β7 integrins are required to give rise to gut-associated CD11c phagocytes with tolerogenic potential
Since β7 integrins were required to efficiently reconstitute ALDE+DC, we next investigated whether β7 integrins are required in CD11c mononuclear cells to generate TREG in vivo. We transferred a 1:2 ratio of BM cells from CD11c-DTR (CD45.1/2) and from either wt (CD45.2) or β7−/− (CD45.2) mice into irradiated recipients (CD45.1) (figure 6A). Eight weeks later we treated the CD11c-DTR:β7−/− chimeric mice with DT, hence depleting DTR+CD11c+ mononuclear cells (CD45.1/2) and leaving mostly β7−/−CD11c+ cells (CD45.2) in all tissues, including MLN (figure 6A, upper dot plots). This strategy resulted in a marked decrease in ALDE+ MLN-DC (figure 6A, lower dot plots), whereas these cells were preserved in DT-treated CD11cDTR:wt control chimeras (figure 6B), hence confirming that BM precursors need α4β7 to give rise to RA-producing CD11c MLN-DC. Next, we assessed the generation of antigen-specific TREG on oral immunisation. Naïve OT-II CD4 T cells were adoptively transferred into CD11c-DTR:β7−/− or CD11c-DTR:wt chimeric mice, which were then immunised with 50 mg ovalbumin (OVA) via oral gavage. Then the mice were treated with DT, as described above. While both sets of untreated mice gave rise to similar proportions of TREG on OVA immunisation, DT treatment resulted in decreased generation of TREG in CD11c-DTR:β7−/−, but not in control CD11c-DTR:wt mice (figure 6B). Of note, all conditions showed similar CFSE-dilution rates on transferred OT-II cells at the time of the analysis (data not shown), suggesting that T cell activation/proliferation was preserved in each case.

Bone marrow (BM) progenitors require β7 integrins to give rise to CD11c mononuclear phagocytes with tolerogenic potential. BM cells from CD11c-DTR (CD45.1/2) and from either wt or β7−/− mice (CD45.2) were transferred in a 1:2 ratio into lethally irradiated recipients. Eight weeks later the mice were treated or not with diphtheria toxin (DT), hence resulting in mice with either wt or β7−/− dendritic cells (DC). (A and B) mesenteric lymph nodes (MLN) cells were analysed for RALDH activity in CD11c DC (n=3). (C) OT-II CD4 T cells were transferred into chimeric mice that were treated or not with DT. Then the mice were orally immunised with ovalbumin (OVA) and three days later the percentages of Foxp3+CD4+ OT-II cells were analysed in MLN (n=3). (D) Chimeric mice treated or not with DT were supplemented daily with oral OVA or PBS for 5 days, and then immunised subcutaneously with OVA plus CFA. After 7 days splenocytes were stimulated with OVA (1 mg/mL) and IL-10 was measured in the culture supernatant after 48 h (n=6 mice/group; three independent experiments). (E) Body weight loss on TNBS-induced colitis in chimeric mice treated every other day with DT (n=14–17 mice/group). (F and G) Flow cytometry analysis for CD4 and Foxp3 expression in cell suspensions from spleen and colon lamina propria from chimeric mice described in ‘E’. Mean±SEM, *p<0.05, **p<0.01. (A, B), unpaired t test; C, ANOVA versus respective first column controls.
We then investigated induction of IL-10 producing T cells on antigen feeding, which relies on the induction of gut-homing CD4 T cells.7 ,8 CD11c-DTR:β7−/− or CD11c-DTR:wt chimeric mice were fed with 2.5 mg of OVA for 5 consecutive days, then immunised with OVA/CFA subcutaneously; 1 week later the splenocytes were analysed for the presence of IL-10-producing cells, as described previously.8 As expected, DT treatment did not affect IL-10-producing cells in CD11c-DTR:wt control chimeras on oral OVA administration (figure 6D, black bars). On the other hand, CD11c-DTR:β7−/− chimeras showed a lower induction of IL-10-producing cells on oral OVA feeding when compared to control chimeras, which was completely abolished on DT treatment (figure 6D, white bars). As CD4 T cells are required for acute TNBS-induced colitis, we chose this model to investigate whether β7 integrins are required on DC to generate Foxp3+ TREG during acute intestinal inflammation. Specific depletion of β7+ DC in CD11c-DTR:β7−/− chimeric mice showed similar body weight loss compared to control CD11c-DTR:wt chimeras (figure 6E). However, the frequencies of colonic Foxp3 TREG were significantly lower in CD11c-DTR:β7−/− when compared to CD11c-DTR:wt chimeric mice (figure 6F–G). These results indicate that β7−/− BM cells are impaired in generating CD11c mononuclear phagocytes with the ability to induce tolerogenic TREG on oral Ag administration.
Discussion
Our data support a model in which BM progenitors acquire the gut-homing receptor α4β7 in a RA-dependent manner (presumably in the BM environment), conferring BM precursors with the capacity to home to the SI-LP and give rise to mononuclear phagocytes, in particular RA-producing ALDE+CD103+ DC, which are required for inducing tolerogenic immune responses (figure 7).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Bone marrow (BM) progenitors require β7 integrins to give rise to gut-associated mononuclear phagocytes with tolerogenic potential. Retinoic acid (RA) (from Aldh1a1+ haematopoietic stem cells? (HSC?)) induces α4β7 on LinnegLy6Clow BM progenitors, which then exit the BM to reach the small intestine lamina propria, a process that requires α4β7. Once in the small intestine, dendritic cells (DC)/Mφ precursors give rise to RA-producing (ALDE+) DC, ALDEneg DC and CD64+ Mφ. After antigen (Ag)-sampling ALDE+DC migrate to the mesenteric lymph node (MLN) in a CCR7-dependent manner, where they induce α4β7+CCR9+ Foxp3+ TREG, a critical initial step for inducing tolerogenic immune responses.
In agreement with a previous report,18 we showed a decrease in CD103 and ALDE+DC in the SI-LP of colitic mice. Interestingly, lack of β7 integrins in the innate immune compartment accelerated inflammation in the same T cell-dependent colitis model, which correlated with a decrease in the numbers of ALDE+DC. This is consistent with our model in which lack of α4β7 impairs the generation of RA-producing DC in the LP, without significantly affecting Mφ, hence potentially disrupting the balance between tolerogenic and pro-inflammatory cell populations. In agreement with this interpretation, we showed that lack of β7 integrins in DC impairs the induction of Foxp3+ and IL-10 producing TREG in vivo.
The requirement for β7 integrins in gut-associated mononuclear phagocyte reconstitution suggests that LinnegLy6Clow precursors need α4β7 to give rise to gut-associated DC. Although we cannot formally exclude that αE(CD103)β7 integrin might also play a role in this process (CD103−/− mice are not commercially available), αEβ7 has not been shown to mediate homing to the gut mucosa.30 ,31 Moreover, CD103−/− mice do not have a defect in DC subsets or in the generation of RA-producing (gut-homing imprinting) DC.21 Therefore, αEβ7 is unlikely to play a role in the recruitment of mononuclear phagocyte precursors to the intestinal mucosa or in their differentiation into RA-producing cells.
Our data favour a model in which undifferentiated precursors, rather than terminally differentiated CD11c cells, are deployed early on to the intestinal mucosa and then proliferate and differentiate into CD11c mononuclear phagocytes.11 ,25 This interpretation is supported by our experimental protocol used to transfer LinnegLy6Clow cells, in which we included anti-CD11c in the lineage cocktail to exclude terminally differentiated CD11chi cells among our transferred BM cells. Given this technical approach, we might have excluded pre-DC (CD11cint) from the LinnegLy6Clow population. Thus, we cannot rule out a role for α4β7 in a more differentiated pre-DC population, as recently suggested.32
Our data suggest that α4β7 targets migratory precursors to the SI-LP to differentiate into ALDE+DC. We observed that wt LinnegLy6Clow BM cells were more efficient to reconstitute DC and pDC in SI-LP when compared to β7−/−LinnegLy6Clow BM cells, which is in agreement with a recent report describing a population of α4β7+CD11cintB220+ BM cells, which give rise to CD103 DC and pDC.32 Interestingly, whereas our data clearly support a role for β7 integrins in generating SI-LP DC and pDC, CCR9 appears to be dispensable, which is somewhat unexpected given previous reports showing that BM-derived DC can be induced to express CCR9 in a RA-dependent manner33 and that CCR9 is required for pDC homing to the small intestine.34 ,35 While the reasons for this apparent discrepancy remain to be determined, the recently described α4β7+CD11cintB220+ BM population lacks CCR9,32 supporting the notion that this receptor is not required for gut DC reconstitution. Of note, reconstitution of colon DC appears to be independent of β7-integrins, which correlates with the low frequencies of ALDE+DC in this gut compartment.12 ,36 Interestingly, LinnegLy6Clow BM cells can rapidly be mobilised on an oral inflammatory stimulus such as cholera toxin (EJV and JRM, unpublished observation), suggesting that gut-tropic LinnegLy6Clow cells might be well poised to quickly replenish ALDE+DC in the intestinal mucosa in the setting of inflammation. This might be the case during T cell-mediated colitis, where ALDE+DC frequencies are decreased after CD4 T cell transfer into β7−/−RAG−/− when compared to RAG−/− mice, whereas in the steady state condition ALDE+DC do not appear to be affected. It is possible that compensatory mechanisms decrease the requirement of β7 integrins in gut DC reconstitution during the steady state, whereas during inflammation the α4β7/MAdCAM-1 homing pathway might become important for the rapid recruitment of DC precursors to the intestinal mucosa. Nonetheless, the advantage of wt over β7−/− DC precursors becomes evident under competitive conditions, such as in our mixed BM chimera experiments.
Although DC probably acquire RA-producing capacity in the SI-LP,12 ,27 ,28 our data show that LinnegLy6Clow cells express α4β7 already in the BM, and that this integrin is required to confer BM DC precursors with the ability to migrate to the intestinal mucosa and differentiate into RA-producing DC. Interestingly, VAD mice (lacking the substrate to synthesise RA) also exhibited a slight but significant decrease in the proportion of BM CDP, which was mirrored by a comparable decrease in the percentages of CD103 cells in MLN and PP. Thus, RA might be partially involved in the generation or expansion/survival of intestinal DC precursors, which is in agreement with a proposed role of RA in HSC differentiation to haematopoietic lineages.37
We and others have previously shown that TREG need to home to the SI-LP for further expansion and differentiation into IL-10 producing cells, which can later be detected outside the gut.7 ,8 It has been proposed that IL-10-producing CX3CR1hi macrophages are necessary to expand and imprint IL-10-producing capacity on TREG in the SI-LP.7 ,8 In this regard, our data show that β7−/− MLN-DC exhibit lower RA-synthesising capacity when compared to their wt counterpart, which correlates with decreased TREG induction in MLN and a lower induction of IL-10-producing T cells. Thus, RA-producing MLN-DC provide the necessary first step to generate gut-tropic TREG, but the final immunogenic or tolerogenic outcome will be decided in subsequent steps taking place in the SI-LP. From a teleological standpoint, it might be advantageous to have the SI-LP as an additional checkpoint to reassess the nature of the antigen, before deciding whether an immune response should be pro-inflammatory/immunogenic or tolerogenic. In this context, our data indicate that BM DC precursors require β7 integrins to give rise to RA-producing gut-associated DC, which are critical for the initial phase of tolerogenic immune responses in the gut mucosa.
Acknowledgments
We thank Jeffrey Lian, Lindsay Kua, and Camilla Engblom for critical reading of the manuscript. JRM is indebted to Ingrid Ramos for constant support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors JDC, PTP and BC designed and performed experiments. DDN analysed and scored histological sections. SG provided valuable suggestions and advice on the manuscript. EJV and JRM conceived and developed the conceptual framework for the study, supervised the project, designed experiments, analysed the data and wrote the paper.
-
Funding EJV was supported by grants from the Crohn's & Colitis Foundation of America (CCFA). JRM was supported by grants from CCFA and NIH DP2 2009A054301.
-
Competing interests None.
-
Ethics approval Mice were maintained in SPF/VAF conditions and used in accordance with the guidelines from the Subcommittee on Research Animal Care at Massachusetts General Hospital and Harvard Medical School.
-
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Commentary