Article Text

Statistics from Altmetric.com
- innate immunity
- complement evasion
- streptococcal inhibitor of complement
- secretory leucocyte protease inhibitor
- BSA, bovine serum albumin
- CCP, complement control protein
- GAS, group A streptococci
- HEL, hen egg lysozyme
- MAC, membrane attack complex
- SCR, short consensus repeat
- SIC, streprococcal inhibitor of complement
- SLPI, secretory leucocyte protease inhibitor
A microbe becomes a pathogen by successfully evading the host’s immune responses, and the microbial strategies for so doing are legion. They include methods to avoid recognition by the immune system—for example, by antigenic variation as shown by influenza and HIV and by parasites or plasmodia, or by acquiring a host coat as is done by worms and retroviruses.
Another major mechanism is to avoid the effector mechanisms of the immune response. This can be done by subverting cytotoxic T cells by the production of decoy HLA molecules; or by subverting Fc function by producing Fc receptor homologues; or by subverting complement by producing homologues of complement control proteins (CCPs). Some viruses also have developed methods of subverting apoptosis in the cells that they infect.
This paper concentrates on the innate immune response. The definition of this term is a little fuzzy. Fearon and Locksley regard all mechanisms using germline coded molecules as being innate which therefore includes natural antibodies with germline V regions.1 Perhaps a more conventional definition is that innate mechanisms are those that are not specifically altered by prior exposure to the same pathogen.
In the first part of this paper some examples of subversion of the complement system will be described. The second half describes some much newer work from our laboratory that takes us into aspects of the innate immune response on mucosal surfaces that have not so far been described.
OVERVIEW OF THE COMPLEMENT SYSTEM
Figure 1 shows a greatly simplified view of complement activation by micro-organisms. Regulation occurs principally at two points—the first is action of the C3 converting enzymes and the second, action of the membrane attack complex (MAC).

Principal activities of the complement system.
Regulation of the C3 converting enzymes is produced by a number of proteins (fig 2), all of which are based on a single protein domain, the so-called CCPs or CCP domain, also called the short consensus repeat (SCR). These proteins are also all located mainly on the long arm of chromosome 1 at a single large locus.
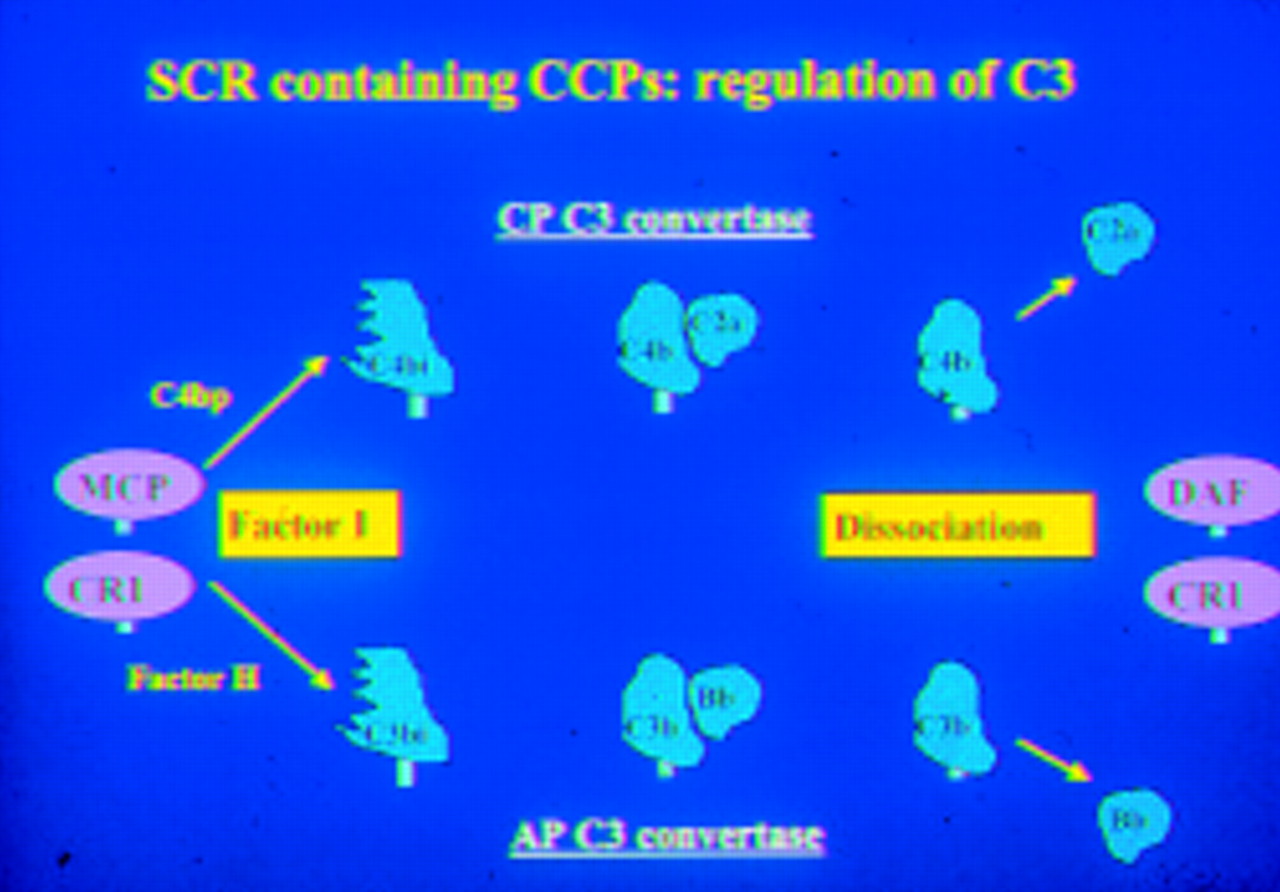
Regulation of C3 activation by proteins containing the complement SCR.
Regulation of the MAC (fig 3) is brought about by a distinctly different protein, CD59. This protein is the only one of its kind in the complement system and similar proteins are not commonly found elsewhere, although its 3D structure (fig 4) does resemble some snake venoms in shape. It is GPI anchored to cell membranes, widely distributed, and important in protecting cells from complement mediated lysis.
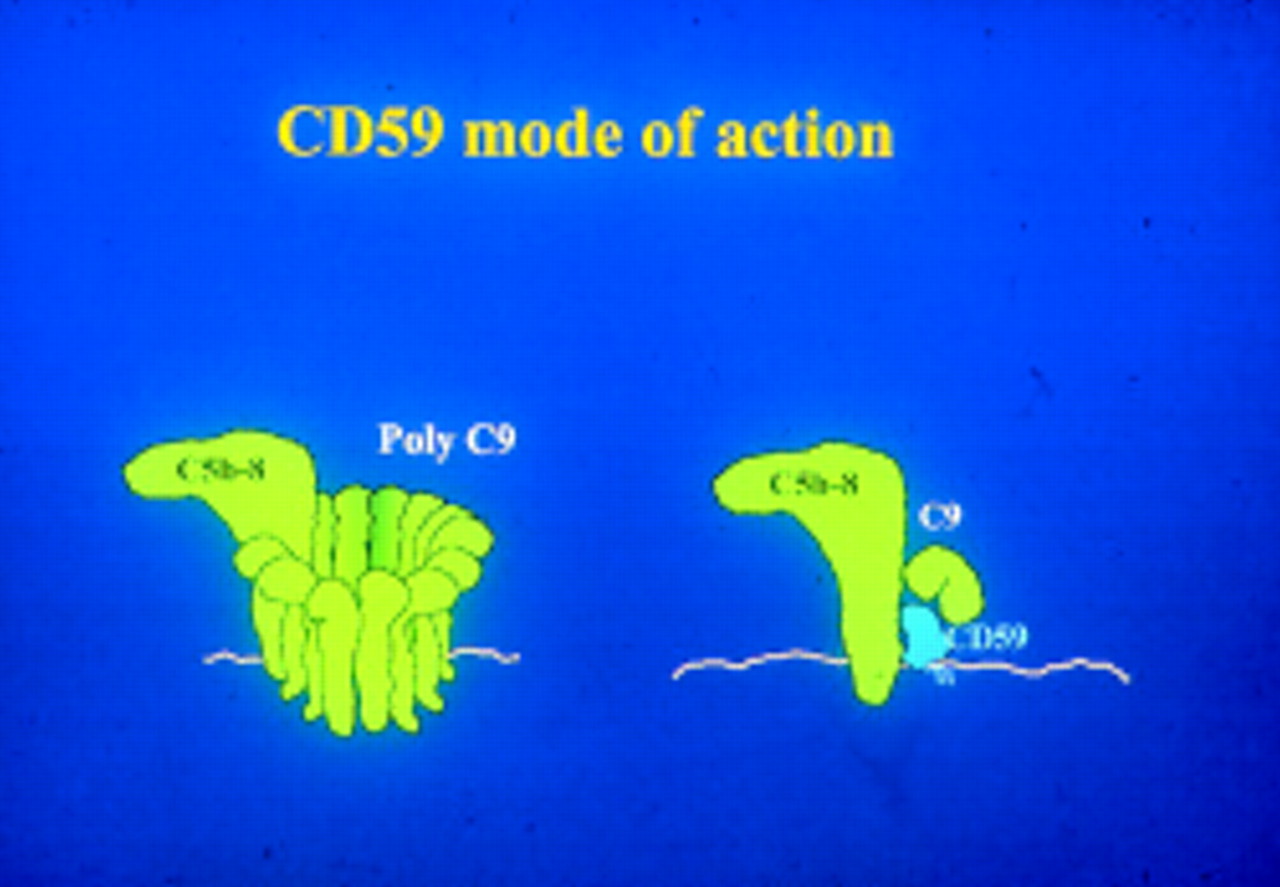
The activity of CD59 in inhibiting the MAC.

The lowest energy structure of CD59. Reproduced from Fletcher CM, Harrison RA, Lachmann PJ, Neuhaus D. Structure of a soluble, glycosylated form of the human complement regulatory protein CD59. Structure 1994;2:185–9. Copyright (1994), with permission from Elsevier Science.
MECHANISMS OF COMPLEMENT EVASION BY VIRUSES
The sources for this section are contained in the review by Lachmann and Davies2.
Viruses have evolved a number of techniques for evading the complement system. One is the arrangement of epitopes in their surface proteins such that antibodies against them are not able to initiate complement fixation. The measles virus provides a good example of this strategy. Antibodies to the measles haemagglutinin, whether IgG or IgM, fail to activate complement by the classical pathway, presumably because the antigenic epitopes are so spaced that effective bridging cannot be obtained between them. This does not prevent measles virus infected cells (even in the absence of antibody) from activating the alternative pathway, but this is a relatively inefficient way of producing cell lysis.
Viruses (for example, HIV) have evolved mechanisms of entry into cells through complement receptors after fixing complement. This is analogous to the strategy used by the Dengue virus which can enter cells through Fc receptors after having bound antibody.
Viruses have also evolved means of acquiring host CCPs. Some viruses—for example, HIV and vaccinia, capture these proteins from the host cell membrane as they leave the cell. Other viruses cause the cell to synthesise specific molecules for this purpose. This in turn can be done in two different ways. One is by gene capture from the host. This will be discussed in relation to herpes virus saimiri. The other is by convergent evolution, where proteins with no structural similarity to host CCPs have evolved that complement control function. This is clearly seen in herpes simplex virus .
Herpes virus saimiri is a T lymphotrophic tumour virus, which harmlessly infects New World primates but in Old World primates gives rise to tumours. Gene sequencing has identified two open reading frames encoding CCPs. One is a homologue of mammalian CCPs with particular homology to C4 binding protein. This protein is produced in two forms: an unspliced, transmembrane protein and a secreted protein produced by alternative splicing. Antibodies against both forms of this protein will neutralise the virus. More unusually, herpes virus saimiri also contains a homologue of CD59 which shows close similarity to human CD59 and, interestingly, an even greater similarity to squirrel monkey CD59. The squirrel monkey is the virus’s natural host and this suggests strongly that the viral CD59 gene has been captured from the squirrel monkey in relatively recent times.
When expressed in insect cells using a baculovirus system the herpes virus saimiri CD59 can be shown to have MAC inhibiting activity. In inhibiting lysis by human serum, viral CD59 is much less efficient than human CD59 but approaches the activity of human CD59 when marmoset serum is used. On the other hand, when tested against lysis by rat serum it had the same activity as human serum; and using rabbit serum, where human CD59 is extremely inefficient, the viral CD59 gives a much stronger MAC inhibiting effect. These data are interesting in that they will assist in mapping the essential residues of CD59 that determine the specificity of reaction with different species of MAC.
Herpes simplex virus has a CCP—G-c1 in HSV1 and G-c2 in HSV2—which has no structural similarity to CCPs. G-c 1 and 2, however, do control C3 convertases, but their activity is slightly different from that of a typical mammalian CCP. Nevertheless, it is known that they are effective in vivo and that their absence reduces viral virulence.
CCPs also occur on a number of parasites. For example Trypanosoma cruzi has a molecule that has decay accelerating factor-like structures and controls C3 convertases.3Schistosoma mansoni has a protein known as SCIP-1, which has CD59-like properties and shows some cross reactivity with antibodies to human CD59.4 SCIP-1 is made only in the mammalian host and not in the snail part of the parasite’s life cycle where resistance to mammalian complement is not needed.
Bacteria do things rather differently. A number of bacteria bind the soluble mammalian CCPs, factor H, and C4 binding protein onto their surface as CCPs. These include Neisseria gonorrhoeae5,6 and Streptococcus pyogenes.7 However, in 1996, Åkesson and his colleagues described a novel protein which is secreted by M1 group A streptococci (GAS) and which did appear to act as a CCP.8 It is this protein that is the topic of the second half of this paper.
STREPTOCOCCAL INHIBITOR OF COMPLEMENT (SIC)
SIC is a 31 kDa extracellular protein with no obvious resemblance to mammalian CCPs and is secreted at high concentrations, about 5 mg/l. It has been shown to inhibit complement lysis. It has also been reported to be highly polymorphic in different GAS and has been used for epidemiological studies.
Lukomski and colleagues in 2000 produced an SIC knockout group A streptococcus and showed that this failed to colonise the throat efficiently.9 We were interested by this report and looked in more detail at its anticomplementary activity. SIC was found to be a C567 uptake inhibitor,10 a property shared by a number of other proteins including clusterin and vitronectin. This form of complement inhibition is not generally regarded as being of major biological importance, largely because the physiological C567 uptake inhibitor is C8, which provides some 70% of all C567 uptake inhibition. It therefore seems implausible that inhibition of C567 uptake is the way that SIC facilitates throat colonisation by streptococci. There are three further reasons for doubt. A secreted C567 inhibitor would be ineffective because such inhibition is required on the cell membrane; and there is no complement present on uninflamed mucosal surfaces. Also, GAS are already resistant to lysis by the MAC as their membranes are protected by a thick cell wall and, during exponential growth, a capsule as well. For these reasons we decided to look at the effect of SIC on other components of the innate immune response on airway surfaces.11
There are three major components of this innate immune response. These are lysozyme present at about 1 mg/ml, lactoferrin at a somewhat similar concentration, and secretory leucocyte protease inhibitor (SLPI) present at a lower, but still appreciable, concentration of 10–80 μg/ml. It has been shown that these three components synergise in killing bacteria.12 A number of further components are present at lower concentrations, including elafin, a smaller molecule related to SLPI, LL-37, and the β-defensins.
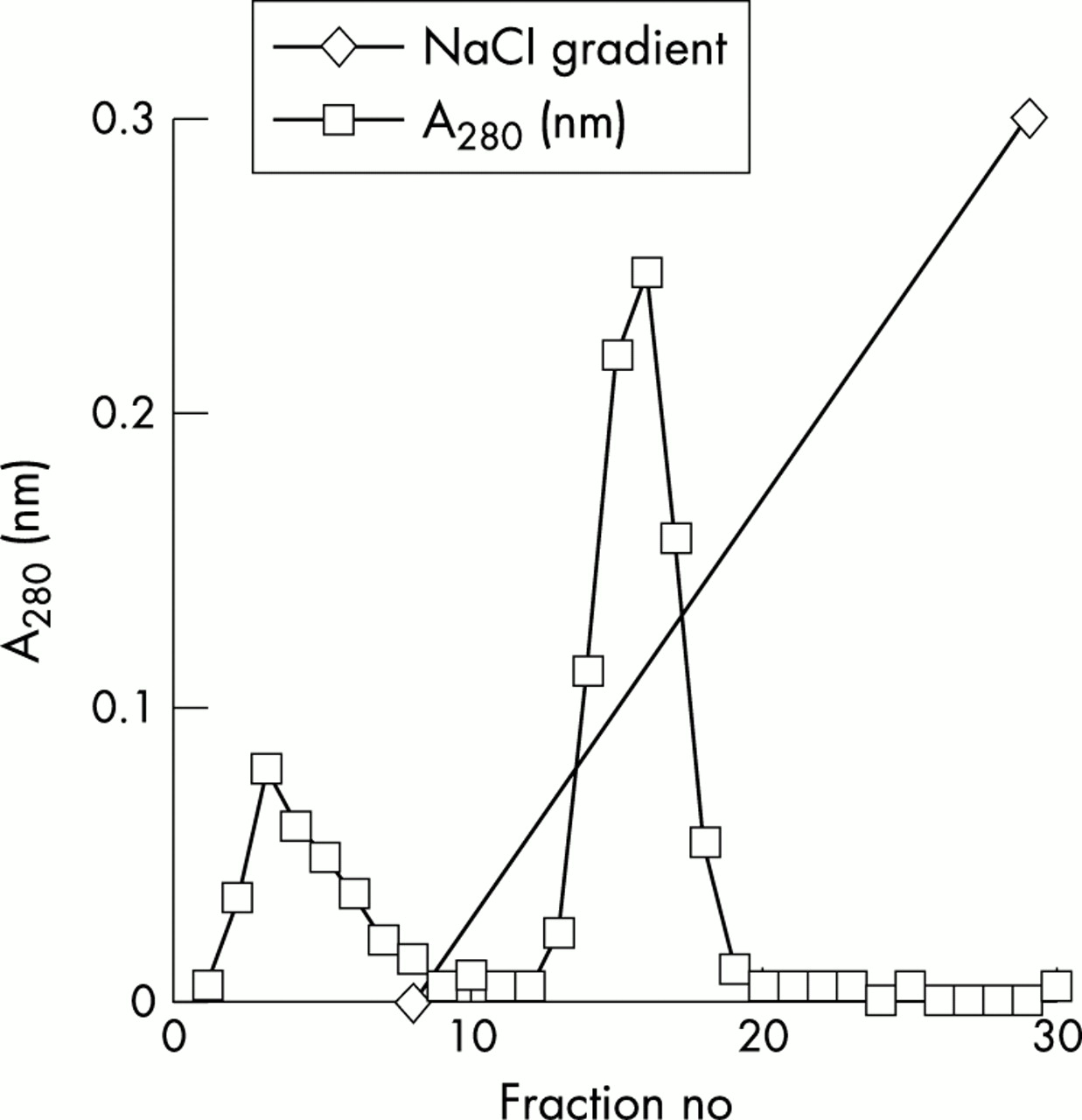
On examination of the three major components we found that on enzyme linked immunosorbent assays (ELISA) SIC binds to both hen egg lysozyme (HEL) and human lysozyme and also to SLPI, but not to lactoferrin. The reaction of lysozyme is impressive in so far as the addition of SIC to lysozyme in solution causes their precipitation (table 1). SIC can be conveniently purified from streptococcal culture supernatants on lysozyme-Sepharose columns at 4°C (fig 5). It is eluted at about 0.3 M NaCl, showing that the binding is fairly strong at that temperature. SIC partially inhibits the lysozyme-induced killing of Streptococcus suis. The inhibition is not complete as S suis are exquisitely sensitive to lysozyme (fig 6). SIC inhibits the catalytic activity of lysozyme in vitro as measured by digestion of bacterial cell walls, the level of inhibition being up to 75%. The reaction is much more efficient at 4°C than it is at 37°C, where indeed it is weak (fig 7).
Precipitation of HEL by SIC at 4°C. Reproduced, with permission, from ref 11. Copyright © 2002 American Society for Microbiology

Affinity purification of SIC on a lysozyme-Sepharose column. HEL was coupled to cyanogen bromide activated Sepharose CL4B to make an 8 ml affinity column and equilibrated with 50 mM Tris/10 mM EDTA pH 7.5 (TE). A 30% ammonium sulphate cut from a 1 litre M1 GAS overnight culture supernatant (resuspended and dialysed into TE) was applied to the column and eluted in a 20 column volume sodium chloride gradient from zero to 2 M, the column being run at 4°C. SIC eluted at around 0.3 M sodium chloride. Reproduced, with permission, from ref 11. Copyright © 2002 American Society for Microbiology.

Inhibition of lysozyme killing of S suis by SIC. Double dilutions of SIC from 17 μM were incubated with 17 μM HEL overnight at 4°C. Equal volumes of S suis at 2.5×109/ml were added together with bovine serum albumin (BSA) to 1 mg/ml and incubated for 15 minutes at 37°C. Final concentrations were bacteria at 109/ml, SIC from 8.5 μM and 8.5 μM HEL. Controls were bacteria in 8.5 μM HEL, dilutions of SIC, or buffer. Tenfold dilutions of the bacteria were plated. Results are expressed as mean (SEM) percentage survival compared with bacteria incubated in buffer only. Reproduced, with permission, from ref 11. Copyright © 2002 American Society for Microbiology.

Inhibition of the catalytic activity of HEL by SIC. Three in four dilutions of SIC from 16.1 μM were combined with 17.5 μM HEL and incubated overnight at 4°C or 37°C. Residual lysozyme activity was assayed by the lysoplate method. Data are from three experiments performed in duplicate and are expressed as mean (SEM) percentage of lysozyme activity remaining compared with controls of lysozyme alone. Reproduced, with permission, from ref 11. Copyright © 2002 American Society for Microbiology.
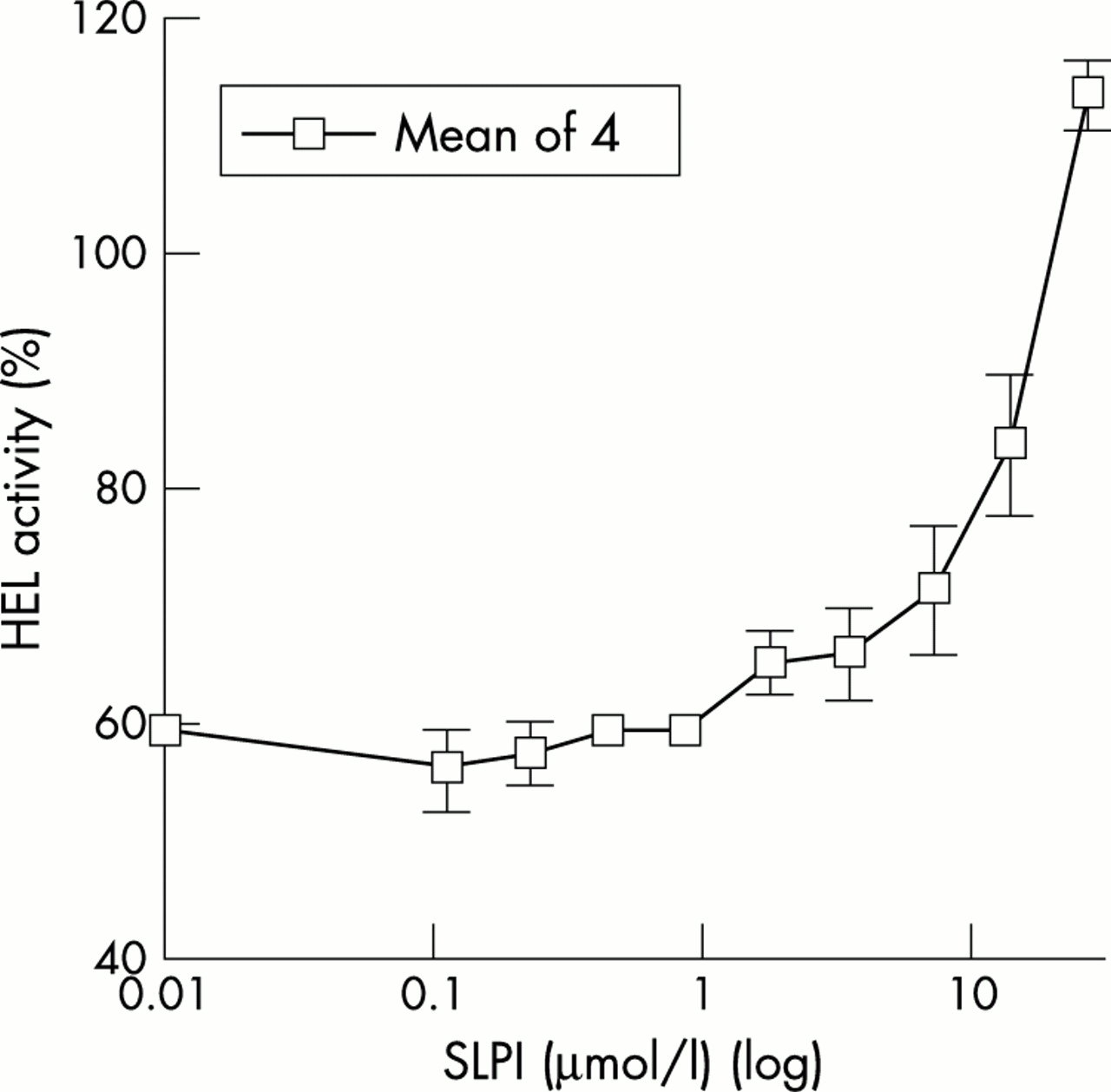
SLPI is a peptide of 11.7 kDa, widely distributed in mucous fluids. Its crystal structure is known and shows two similar domains. The C-terminal domain has protease inhibitory activity and the N-terminal domain has bactericidal activity. SLPI does kill the M1 strain of GAS, which lysozyme does not, and interestingly this activity is inhibited by SIC (fig 8). On the other hand, SIC has no effect on elastase inhibition by SLPI.

SLPI kills the M1 strain of GAS and killing is inhibited by SIC. (A) Doubling dilutions of SLPI from 20 μM were combined with bacteria at 105/ml together with BSA at 1 mg/ml and incubated for two hours at 37°C. Tenfold dilutions of the bacteria were plated. Results are from 2 experiments performed in duplicate. (B) Doubling dilutions of SIC from 40 μM were incubated with 40 μM SLPI for two hours at 37°C. Bacteria at 2.5×105/ml were added together with BSA to 1 mg/ml, and incubated for a further two hours at 37°C and processed as above. Final concentrations were bacteria at 105/ml, SIC from 10 μM, and 10 μM SLPI. Controls were bacteria in 10 μM SLPI, buffer, or dilutions of SIC. Results are from a typical experiment performed in duplicate. In (A) and (B) results are expressed as mean (SEM) percentage survival compared with bacteria incubated in buffer only. Reproduced, with permission, from ref 11. Copyright © 2002 American Society for Microbiology.
Competition experiments suggest that SLPI and lysozymes bind closely to each other on SIC (fig 9). However, we do not believe that the binding sites are identical because the properties of the binding are very different. The reaction of SLPI with SIC shows a slight paradoxical temperature dependence, occurring better at 37°C than 4°C. This shows that the binding is largely hydrophobic. The maximum number of SLPI molecules bound to each SIC molecule is probably two. On the other hand the binding of lysozyme is extremely temperature dependent and occurs effectively only at 4°C, showing the reaction to be largely ionic. The maximum number of molecules bound is probably four (table 2).
Binding characteristics of SIC with SLPI and HEL. Reproduced, with permission, from ref 11. Copyright © 2002 American Society for Microbiology.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Competition between SLPI and HEL for binding to SIC. Double dilutions of SLPI from 28.5 μM were incubated with 10 μM HEL and 5 μM SIC. HEL activity released (and hence the degree of competition by SLPI for binding to SIC) was assayed by the lysoplate method. Results are expressed as mean (SEM) percentage of lysozyme activity compared with control of 10 μM lysozyme alone and represent four experiments performed in duplicate. Reproduced, with permission, from ref 11. Copyright © 2002 American Society for Microbiology.
Thermodynamic calculations confirm that the reaction with SLPI is largely entropy driven and has very small positive enthalpy, whereas the reaction with lysozyme has a very small entropy change and a substantial negative enthalpy. At 4°C the free energy changes are quite similar (table 3). These data suggest that the binding sites are different, although close to each other. They also suggest that the in vivo role of SIC is likely to lie predominantly in its binding with SLPI.11
Physical chemical data—interaction of SIC with SLPI and HEL. Reproduced, with permission, from ref 11. Copyright © 2002 American Society for Microbiology.
SIC is, to the best of our knowledge, the first biological inhibitor of SLPI which has been described. It is clearly a target for developing antistreptococcal treatments. It is also likely to prove a useful reagent in probing in vivo activities of SLPI and lysozyme in both infective and uninfective models of mucosal inflammation.