Article Text

Statistics from Altmetric.com
SUMMARY
The nervous system in the intestine controls motility, secretion, sensory perception, and immune function. Peptidergic neurones with neurotransmitters such as substance P and nerve growth factors have been the main focus of neuroimmunomodulation research in the gut. This review summarises the present knowledge concerning the role of the sympathetic nervous system (SNS) in modulating intestinal inflammation. The role of the SNS for gut inflammation is compared with its role in rheumatoid arthritis which demonstrates notable similarities. Nerve fibres of the SNS not only enter the enteric plexuses but also innervate the mucosa and gut associated lymphoid tissue (GALT). The SNS has pro- and anti-inflammatory functions. Neurotransmitters such as norepinephrine, adenosine, and others can evoke remarkably different opposing effects depending on concentration (presence of sympathetic nerve fibres and extent of neurotransmitter release), receptor affinity at different receptor subtypes, expression of adrenoceptors, availability of cotransmitters, and timing of SNS activity in relation to the inflammatory course. This review attempts to integrate the different perspectives of the pro- and anti-inflammatory effects of the SNS on inflammatory disease of the gut.
INTRODUCTION
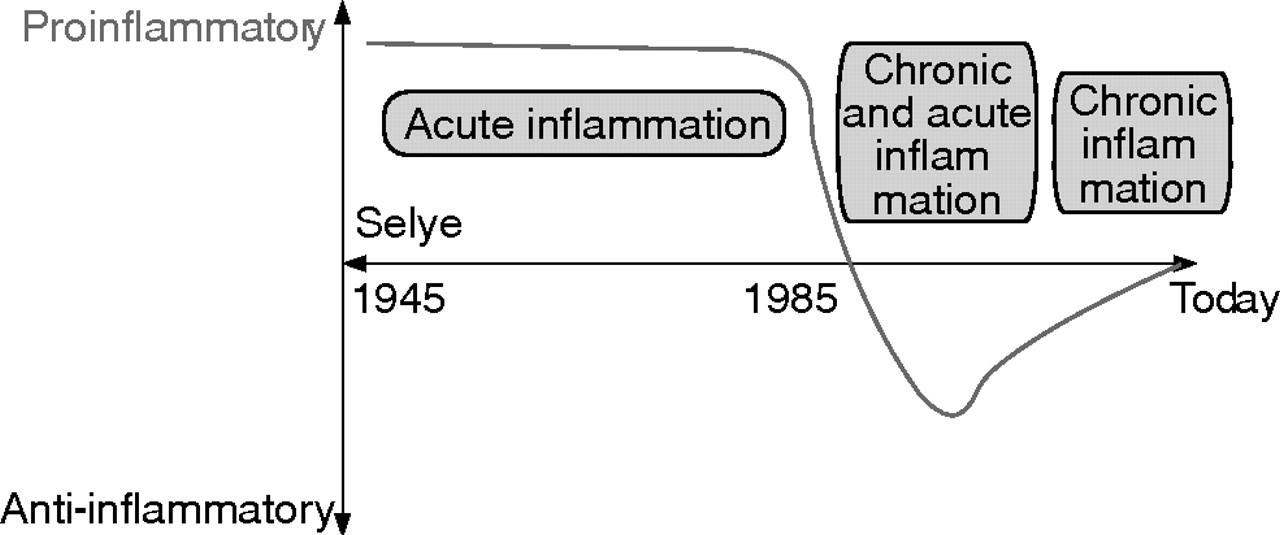
Since the work of Selye in the 1940s, the SNS together with the hypothalamic-pituitary-adrenal (HPA) axis was thought to play an important supportive role in the fight and flight reaction during stressful situations (fig 1⇓).1 In linking this important concept to inflammatory diseases, several groups, from 1960 to the late 1980s, delineated the proinflammatory role of the SNS for the early inflammatory response (fig 1⇓) (for example, see Levine and colleagues2). Indeed, the SNS is a critical proinflammatory component of neurogenic inflammation, which is particularly evident during the first hours of induction of inflammation.3 This is most probably due to the supportive effects of neurotransmitters on plasma extravasation and directed migration of immune cells to sites of inflammation (summarised by Dhabhar and McEwen4).
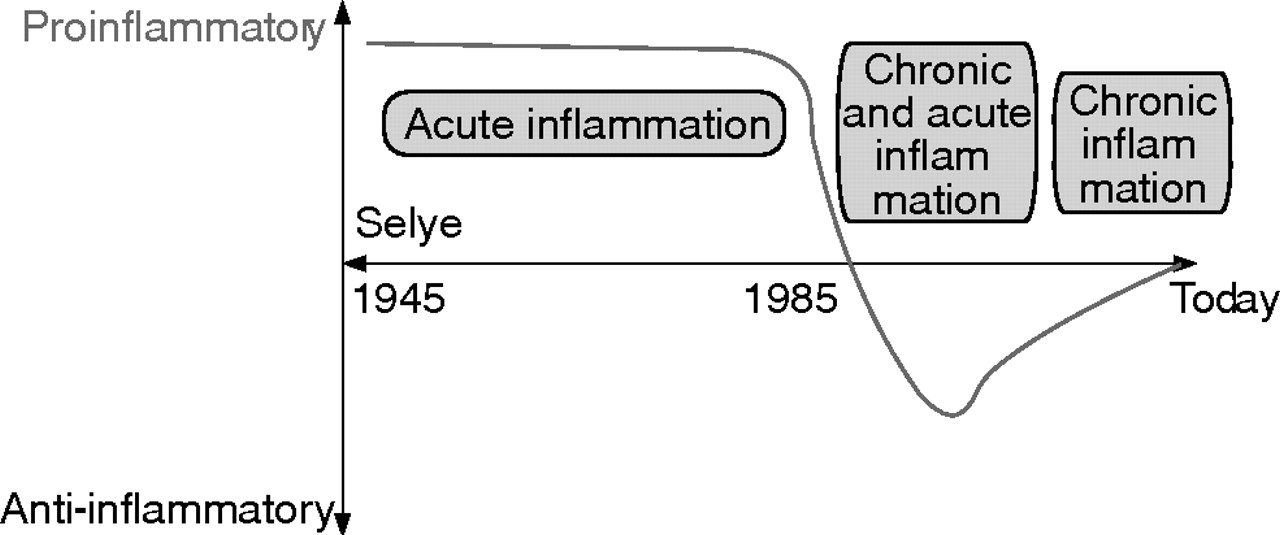
Historical consideration of the sympathetic nervous system (SNS) in relation to inflammation. Between the 1940s and the late 1980s, it was thought that the SNS supports the inflammatory process in the context of a fight and flight reaction. In this period, mainly short lived inflammatory processes were investigated over a few hours. Since about 1985, with recognition of strong inhibitory influences on secretion of immune mediators such as tumour necrosis factor in cell culture experiments, the SNS was thought to play an anti-inflammatory role in arthritis. Nowadays, it is generally agreed that the SNS plays either a pro- or anti-inflammatory role depending on the time point of SNS modulation in relation to inflammation.
At the beginning of the 1980s, with the appearance of in vitro immune cell culture assays for several days and development of immunoassays for the detection of distinct proinflammatory cytokines such as tumour necrosis factor (TNF), interferon γ (IFN-γ), interleukin (IL)-2, or IL-12, important immune inhibitory effects of sympathetic neurotransmitters via β-adrenoceptors and A2 adenosine receptors, via cyclic adenosine monophosphate (cAMP), protein kinase A, and cAMP responsive element binding protein were described in vitro (for example, see Johnson and colleagues,5 Novogrodsky and colleagues,6 Renz and colleagues,7 Snijdewint and colleagues, and 8 Elenkov and colleagues9). In the following years, observations encouraged a new understanding of the role of the SNS as independent authors noted either pro- or anti-inflammatory roles for the SNS, as exemplified in arthritis research.2,3,10–,12 Recent understanding of the SNS in inflammation strongly suggests that both viewpoints are probably correct. This review tries to integrate these different viewpoints of the pro- and anti-inflammatory effects of the SNS in gut inflammation.
ANATOMY AND PHYSIOLOGY OF THE SYMPATHETIC NERVOUS SYSTEM IN THE GUT
Control levels of the sympathetic nervous system
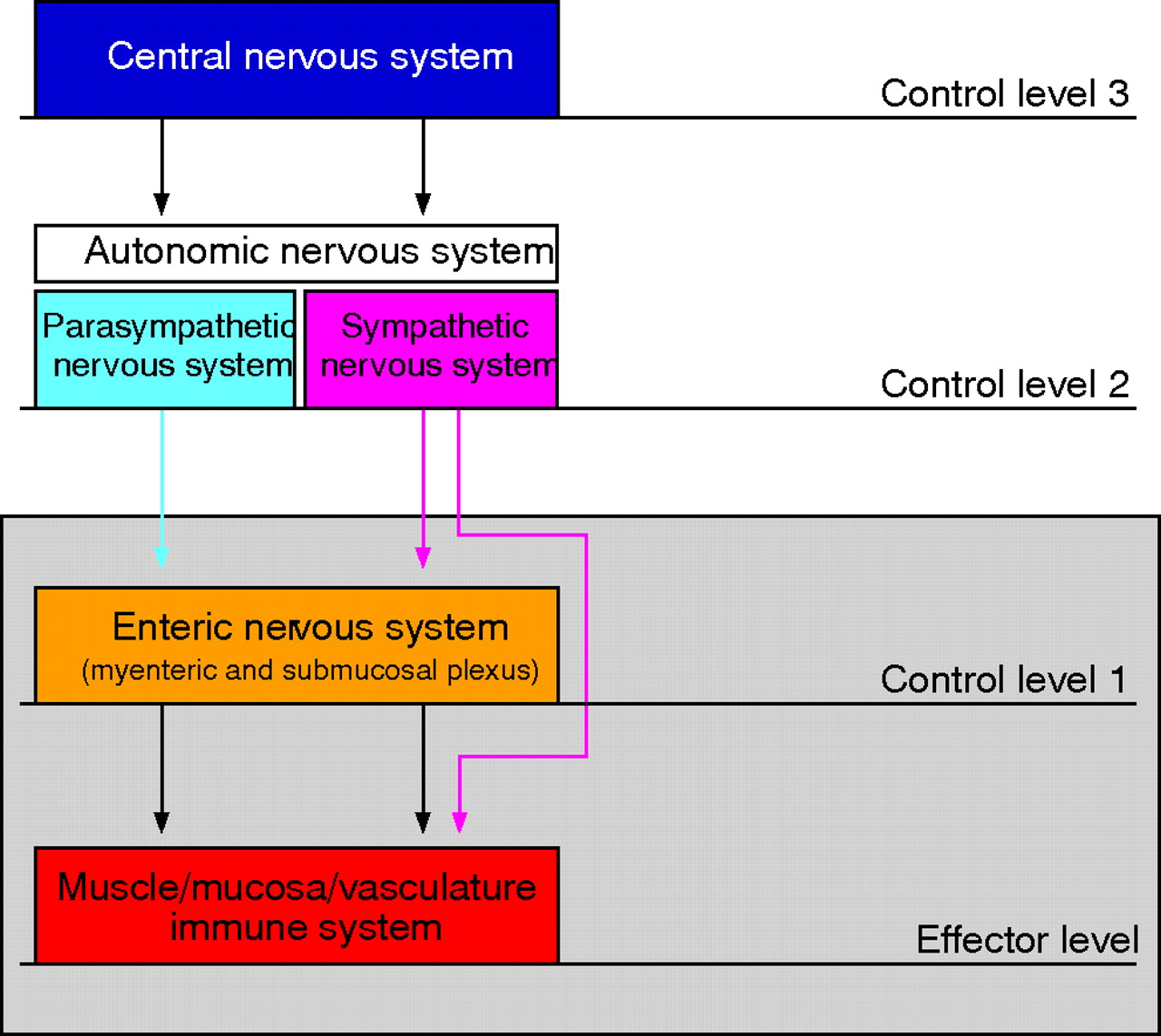
Central sympathetic outflow is controlled by sympathetic control centres in the brain which are activated by either central nervous stimuli (cortical areas, limbic system) or by different inputs from the periphery (via the hypothalamus) or sensory afferent nerves (fig 2⇓).13 Input signals to the brain from the periphery can be circulating cytokines such as IL-1β or stimulation of sensory nerve fibres by cytokines.14–,16 Different brain centres activate efferent sympathetic pathways, which pass through the intermediolateral column of the spinal cord and reach sympathetic ganglia in the proximity of the aorta or in the abdomen. Then, nerve fibres are switched to postganglionic sympathetic noradrenergic nerve fibres, which enter the gut through the mesenteric serosal surface (fig 2⇓). The hierarchy of control centres is mandatory for adequate and site specific control of the SNS on different levels from the brain to the periphery.

Neuronal control of the intestine has a hierarchical organisation with different levels of integrative organisation (adapted from Wood and colleagues. Gut 1999; 45(Suppl II):II6–II16).
Microanatomy of the sympathetic nervous system in the gut
The serosal surface of the intestine is densely innervated by sympathetic nerve fibres.17 In 1965 it was reported that sympathetic nerve fibres are normally infrequent in the intestinal mucosa.18 This view changed during the 1970s and 1980s when improved histological techniques became available.19–,22 Sympathetic nerve fibres enter the intestinal wall along arteries and terminate in the myenteric and submucosal plexuses, and in the mucosa.21,22 It is important to mention that sympathetic nerve fibres not only terminate in vessel walls or enteric plexuses in order to control vascular tone or secretomotor neurones but also end in the larger vicinity of blood vessels in the submucosa and mucosa.21
With respect to the GALT, sympathetic noradrenergic nerve fibres innervate both the vasculature and parenchymal fields of lymphocytes (fig 3⇓).22 This innervation is directed into zones of T lymphocytes and plasma cells.22 In the GALT, represented by the appendix and Peyer’s patches, noradrenergic fibres enter at the serosal surface, travel longitudinally in the serosa and on the submucosal border of the muscularis interna, turn radially into an internodular nerve fibre meshwork, plunge directly through the T cell zones, and ramify profusely among lymphocytes, enterochromaffin cells, and plasma cells in the interdomal regions (fig 3⇓).21 In two studies, it was reported that sympathetic nerve fibres have a similar behaviour in different species, including humans.19,22

Organisation of the sympathetic innervation in gut associated lymphoid tissue (adapted from Felten and colleagues22). Noradrenergic fibres enter the serosal surface (S) pass through the muscle layers (ME, muscularis externa; MI, muscularis interna) and touch the myenteric plexus (MP), form a mesh of sympathetic nerve fibres (SMF) at the submucosal (SM) border of the MI, and then turn radially to run between lymph nodes (N) as internodular nerve fibres (INF). Fibres pass through the T dependent zone (T) and enter the interdomal region (ID) between the domes (D) and then branch profusely in this zone among lymphocytes, enterochromaffin cells, and subepithelial plasma cells. BV, blood vessel; SMP, submucosal plexus.
Neurophysiology of the sympathetic nervous system in the gut
Most often the SNS is linked to the control of motility, secretion, and vasoregulation (fig 4⇓). Through α2 adrenergic inputs, the SNS inhibits postsynaptic potentials of motoneurones in the myenteric plexus and of secretoneurones in the submucosal plexus (fig 4⇓). Furthermore, via α1 adrenergic pathways, the SNS counteracts vasodilation induced by substance P, calcitonin gene related peptide, nitric oxide, and others (fig 4⇓).23 The question arises as to the role of the SNS in the mucosa and GALT. Interestingly, important reviews on the enteric nervous system did not discuss the immunomodulatory role of sympathetic neurotransmitters on mucosal immune cells.23–,25 The role of sympathetic neurotransmitters for immunomodulation is outlined below.
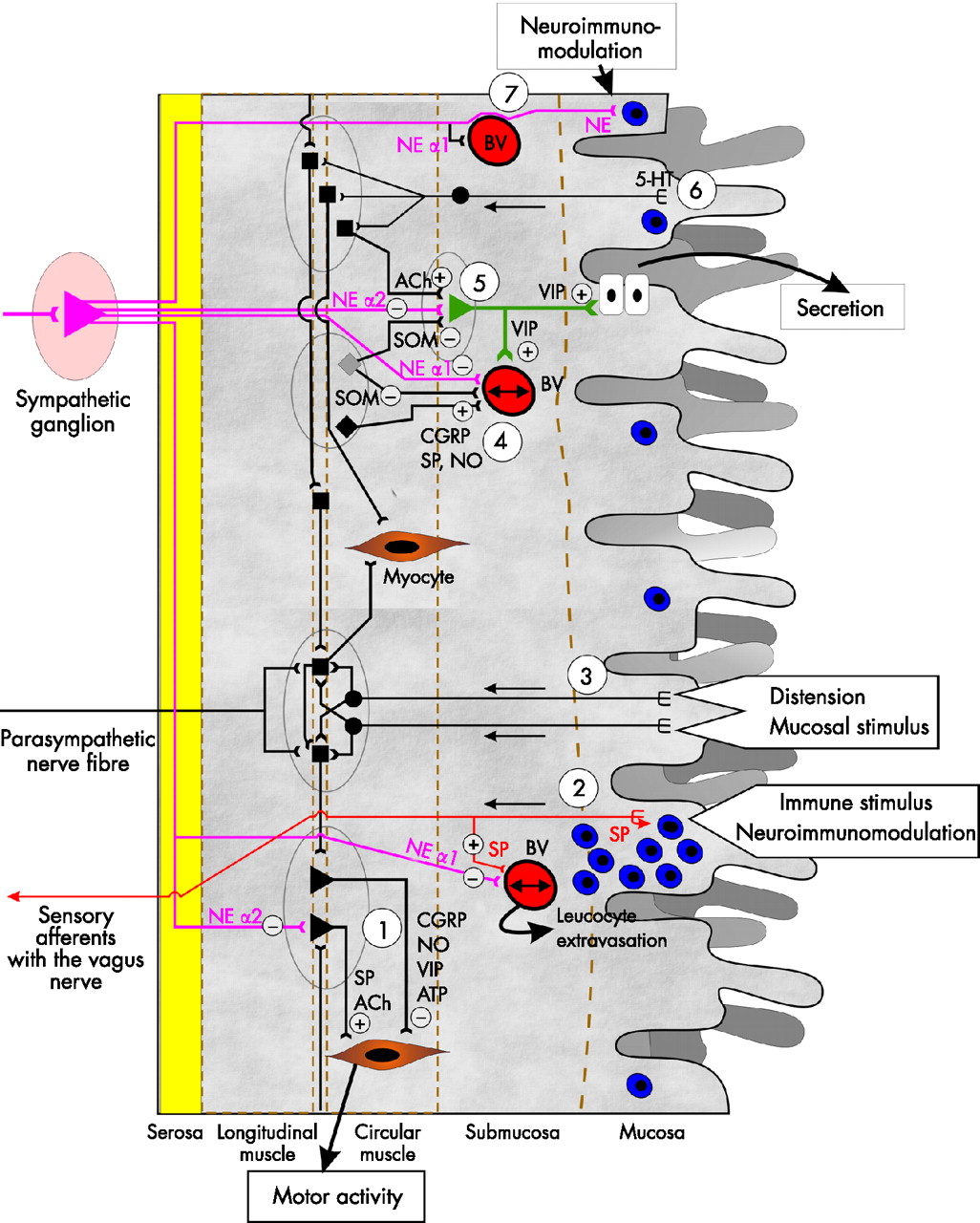
Connections of sympathetic nerve fibres (purple) with structures in the intestinal wall. A plus sign indicates a positive and a minus sign indicates a negative influence. (1) Sympathetic nerve fibres terminate at motoneurones in myenteric plexus ganglia (grey ellipses). Here, norepinephrine (NE) inhibits descending motoneurones (ACh, acetylcholine; SP, substance P) via α2 adrenergic signalling. (2) Immune stimuli from the lumen and in the mucosa stimulate sensory neurones of the vagus, which locally release SP and signal the information to higher centres. SP can attract leucocytes (blue) and can support vasodilation (line with two arrows in the blood vessel (BV)). Via α1 adrenergic receptors, NE leads to vasoconstriction (opposite to SP). (3) Distension and mucosal stimuli can stimulate intrinsic afferent neurones, which modulate motoneurones. (4) Modulation of vascular tone via dilatory signals (SP; CGRP, calcitonin gene related peptide; NO, nitric oxide; VIP, vasoactive intestinal peptide) and constriction signals (SOM, somatostatin; NE via α1 adrenergic receptors). (5) Regulation of the secretory neurone of the submucosal plexus by secretory signals (ACh) and inhibitory signals (NE via α2 adrenoceptors; SOM). (6) Stimulation of intrinsic afferent neurones by serotonin (5-HT). These neurones are coupled to motoneurones and secretory neurones as wells as interneurones in the myenteric plexus. (7) Local efferent sympathetic nerve fibres travel along blood vessels (BV) into the submucosa and mucosa and terminate next to epithelial cells. These efferent fibres can modulate immune responses in the vicinity of mucosal blood vessel. ATP, adenosine triphosphate.
Neurotransmitters of the sympathetic nervous system
Apart from norepinephrine, sympathetic varicosities store neuropeptide Y (NPY), methionine-enkephalin, leucine-enkephalin, β-endorphin, and adenosine triphosphate (ATP) in small and large vesicles.26 These neurotransmitters are co-released.27 It is important to mention that norepinephrine and ATP are locally recycled whereas peptide neurotransmitters are transported along the sympathetic axon to the nerve terminal.26 At a high stimulation frequency over a longer period of time, the sympathetic nerve terminal is devoid of peptide neurotransmitters in relation to norepinephrine and ATP due to the transport and production limit.
Neurotransmitters of the sympathetic nerve terminal are ligands at functionally different receptors with opposing intracellular signalling pathways, which is best elucidated for norepinephrine and adenosine.28 At low concentrations (10−9 to 10−7 M), norepinephrine and adenosine bind to α-adrenoceptors and A1 adenosine receptors, respectively, leading to decreased cAMP levels. At high concentrations (10−7 to 10−5 M), these neurotransmitters bind to β adrenoceptors and A2 adenosine receptors, increasing cAMP levels. Cotransmitters such as NPY support effects via the α or β adrenoceptors depending on the predominant adrenergic signalling pathway.29 The β adrenergic signalling pathway is further supported by cooperative effects of available cortisol, as demonstrated in different cell types.30–,37 Cortisol enters the tissue without restraint. Loss or rapid degradation of endogenous cortisol is most probably a prerequisite for predominant α adrenergic signalling because cortisol exclusively supports the β adrenergic pathway. The cooperative effect has recently been demonstrated in inflamed synovial tissue of patients with rheumatoid arthritis.38
Sympathetic neurotransmitters modulate immune responses
Migration of immune cells
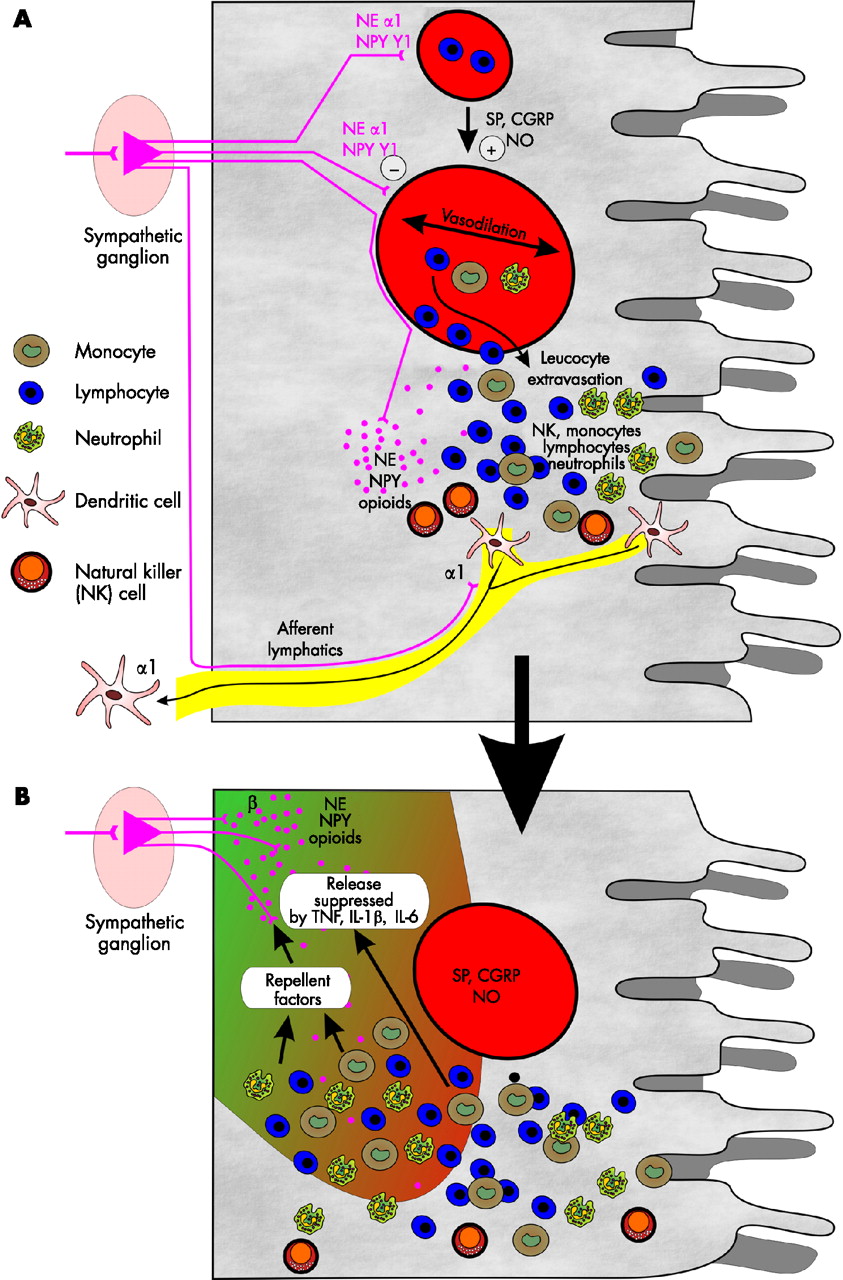
In addition to direct modulation of immune cells (see below), norepinephrine and cotransmitters are important substances for redistribution and directed migration of immune cells (fig 5A⇓).39 Injection of norepinephrine intravenously (as well as cortisol) mobilises many types of immune cells.40 Directed migration of monocytes is partly mediated via β adrenergic signalling and via the NPY Y1 receptor.39 Furthermore, adenosine can attract neutrophils,41 and norepinephrine stimulates secretion of the neutrophil chemotactic cytokine IL-8.42 Instillation of norepinephrine into the intestinal lumen of the mid-ileum increased neutrophil influx to the intestinal mucosa.43 Stress during surgery increased lymphocytes in the lymphatic tissue of the intestine.44 It was further shown that sympathectomy reduced migration of adoptively transferred cells into Peyer patches.45 After resuscitation in a haemorrhagic shock model, leucocyte rolling in gut vessels markedly increased, which was not observed in sympathectomised animals.46 The functional relevance of the SNS for directed migration of leucocytes into dermal tissue has recently been reviewed.4 Interestingly, migration of immature dendritic cells to lymph nodes is mediated via α1 adrenergic receptors, as exemplified in the skin.47 As the SNS responds very rapidly (as a nervous system), these early effects of the SNS are important at the beginning of a local inflammatory tissue response (fig 5A⇓).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Sympathetic influence on vasoregulation, leucocyte extravasation, and dendritic cell exodus. (A) An immune stimulus induces local production of substance P (SP), calcitonin gene related peptide (CGRP), and nitric oxide (NO). Among others, these mediators lead to vasodilation. Norepinephrine (NE) and neuropeptide Y (NPY) counteract vasodilation via α1 adrenergic and NPY Y1 receptors. Chemotactic factors, including SP and NE via β adrenoceptors and sympathetic opioid peptides, support leucocyte extravasation. Elevated concentrations of NE and NPY is indicated by numerous purple dots. Thus neurotransmitters of the sympathetic nervous system support chemotaxis, which is a very early event in the beginning of an inflammatory reaction. In addition, exodus of relatively immature dendritic cells is supported by α1 adrenergic signalling. (B) After leucocytes are encountered in an inflammatory process, they start to produce proinflammatory cytokines and sympathetic nerve repellent factors, which inhibit neurotransmitter release and lead to loss of sympathetic nerve fibres. This development is typical for the early phase of a wound healing process. TNF, tumour necrosis factor; IL, interleukin.
Sympathetic neurotransmitters modulate apoptosis
High concentrations of norepinephrine stimulate apoptosis in different cell types.48–,53 In the intestinal tract, it has been demonstrated that exercise, stress, and catecholamine infusion induced β adrenergically mediated apoptosis of intestinal lymphocytes.54 Induction of apoptosis can be an anti-inflammatory mechanism if proinflammatory immune cells are targeted.
Sympathetic neurotransmitters modulate innate immune cells
Norepinephrine at high concentration (via β adrenoceptors) has been shown to inhibit immune functions such as phagocytosis, natural killer (NK) cell activity, and MHC class II expression, as well as secretion of TNF, IL-12, and IFN-γ from macrophages or lymphocytes (for review see Elenkov and colleagues55). This β adrenergic effect of norepinephrine is supported by an increase in the IκB cytoplasmic half life.56 It is generally accepted that β adrenergic signalling inhibits many aspects of the innate immune system (NK cells, neutrophils, macrophages, and others). However, the effects of norepinephrine at low concentration mediated via α2 adrenoceptors can even increase macrophage TNF secretion.57 Signalling through α2 adrenoceptors can be important to resist the intracellular growth of microbes.58 Similarly, adenosine exerts opposite effects on cytokine secretion at low concentrations compared with high concentrations.59 The dual role of sympathetic neurotransmitters is thus an important prerequisite for either the pro- or anti-inflammatory effects of the SNS on the innate immune system. As mentioned above, the activity of the SNS and presence of neurotransmitters plus their cotransmitters largely define the effects of sympathetic modulation. In addition, regulation of adrenoceptor subtypes on immune cells depending on their state of activation is another mechanism by which sympathetic neurotransmitters are differentially operative (see below).
Sympathetic neurotransmitters modulate cells of the adaptive immune system
Elenkov and colleagues concluded that norepinephrine via β-adrenergic signalling stimulates aspects of T helper type 2 (Th2) immune responses by increasing IL-4, IL-5, IL-6, and IL-10.55 In addition, norepinephrine stimulates immunoglobulin production of B lymphocytes.60 From this point of view, a proinflammatory immune reaction with a predominance of Th2 cytokines such as in ulcerative colitis, systemic lupus erythematosus, or allergic diseases is likely aggravated by the SNS. Interestingly, one study also demonstrated a supportive effect of the β-adrenergic agonist salbutamol on immunological tolerance.61 In contrast, typical T helper type 1 (Th1) immune responses such as production of lymphocyte TNF, IL-2, or IFN-γ are suppressed via the β adrenergic receptor.55 Thus the prevailing type of T lymphocyte reaction determines the influence of the SNS on the immune system. We recently demonstrated such opposite effects of the SNS on immune responses in bacterial infection with Gram negative (TNF is bacteriostatic) and Gram positive (IL-4 is bacteriostatic) bacteria.62
With respect to human immune mediated diseases, the preponderance of a specific B or T lymphocyte immune reaction is often not detectable. A mixture of different types of immune reactions with innate and adaptive (B, Th1, Th2, T regulatory) aspects is present in humans, and the dominant response largely depends on genetic prerequisites of the patient, the antigen, the site of the immune response (co-signals), and the time point of the immune response.
Direction of influence of the sympathetic nervous system depends on the time point of the immune response
In the pre-symptomatic phase of an immune mediated disease, a limited number of cell types are involved. If we accept the aetiological concept of an antigen driven disease (self or foreign), T cells, B cells, and antigen presenting cells will play a major role in the pre-symptomatic phase (as easily recognised in animal models). Recent important articles demonstrated increased autoantibody titres against autoantigens (a function of the adaptive immune system) many years before the first symptoms appeared.63,64 This indicates that immunity is not instantaneously accompanied by symptomatic disease. At a time when the disease becomes symptomatic, many other local cell types, particularly of the innate immune system, are involved in the destructive process. The role of the initial secret players of the adaptive immune system—T cell, B cell, and APC—simultaneously decreases while other cell types are involved. Thus one can separate a pre-symptomatic from a symptomatic phase of the disease. Consequently, the influence of the SNS on the immune response largely depends on the time point of SNS activation in relation to the immune response. Furthermore, we must distinguish the very first phase of an inflammation, during which directed migration is maximally important (supported by the SNS), from later phases of tissue destruction by cells of the innate immune system (inhibited by the SNS). We recently demonstrated that this can largely change the outcome in arthritis in animals.12 The dual role of the SNS depends on the immune mechanisms involved.
THE SYMPATHETIC NERVOUS SYSTEM IN GUT INFLAMMATION
Sympathetic innervation in gut inflammation
Previous studies in arthritis and diabetes research demonstrated extensive loss of sympathetic nerve fibres in the inflamed area.65–,67 Dvorak and Silen demonstrated axonal loss of autonomic nerves in the surgically resected ileum of patients with Crohn’s disease.68 They suggested that affected axons belong to the SNS. Others have demonstrated that gut infection with Toxoplasma gondii resulted in colonic pseudo-obstruction due to selective sympathetic denervation.69 Interestingly, appearance of numerous small TH positive nerve cell bodies and NPY positive nerve fibres were found in clusters in myenteric ganglia in patients with Crohn’s disease but not in control specimens.70 However, sympathetic nerve fibres were rare in the circular muscle layer of the ileum in controls and patients with Crohn’s disease.70 Unfortunately, in this detailed study, sympathetic innervation of the GALT, submucosa, and mucosa was not investigated. In our recent studies in patients with Crohn’s disease there was marked loss of sympathetic nerve fibres in the mucosa and submucosa but not in circular or longitudinal muscle compared with control specimens of tumour patients (F Grum, unpublished results). In addition, we recently demonstrated marked loss of sympathetic nerve fibres in dextran sodium sulphate colitis in mice.71
Geboes reported that the presence of immune cells was associated with alterations of the enteric nervous system.72 This may indicate that immune cells are able to produce factors which influence the plasticity of the local nervous system. In the gastrointestinal tract, neurotrophic factors regulate plasticity of the nervous system.73 However, nerve growth factors are not specific for the different types of nerve fibres, which necessarily does not explain the differential loss of sympathetic nerve fibres during inflammation. In contrast with nerve growth factors, nerve repellent factors are much more specific for nerve fibre repulsion.74–,76 We recently demonstrated that macrophages and fibroblasts in inflammatory lesions produce nerve repellent factors specific for sympathetic but not for sensory nerve fibres.77 One may speculate that during the early process of inflammation the SNS supports directed migration but on activation of local macrophages and fibroblasts, secreted nerve repellent factors lead to distinct loss of sympathetic nerve fibres (fig 5B⇑).
The situation seems to be remarkably different in ulcerative colitis where the density of the adrenergic network was significantly pronounced. However, authors used glyoxylic acid induced fluorescence which is not as specific as antityrosine hydroxylase fluorescence.78
Levels of sympathetic neurotransmitters in intestinal inflammation
As sympathetic nerve fibres are lost in inflamed areas, tissue concentrations of sympathetic neurotransmitters should be decreased. Indeed, in Crohn’s disease, norepinephrine tissue levels in both non-inflamed and inflamed colonic mucosa were markedly lower than in control subjects.79 Similarly, dopamine levels in the inflamed mucosa of Crohn’s disease and ulcerative colitis were markedly lower than in controls whereas L-DOPA levels were elevated.79 As L-DOPA is the precursor of dopamine (and norepinephrine), these findings suggest decreased L-DOPA decarboxylase activity in inflamed tissue.79 Similarly, in the trinitrobenzene sulphonic acid (TNBS) colitis model, dopamine and norepinephrine levels were markedly lower in the inflamed mucosa of the distal colon but not in the non-inflamed ileum.80
A second important factor for low catecholamine levels in inflamed tissue is inhibition of norepinephrine release from sympathetic nerve terminals. This was described by Swain and colleagues in 1991 who demonstrated that intestinal intraepithelial infection with Trichinella spiralis suppressed electrically induced release of norepinephrine. Although the worm infection only lasts 17 days, norepinephrine release was inhibited for over 100 days post infection.81 At the same time, Elenkov and colleagues found that TNF inhibits release of norepinephrine from noradrenergic axon terminals in the isolated rat hypothalamus.82 Rühl and colleagues further corroborated these findings in the intestine using IL-1β and IL-6 and this effect was mediated via induction of nitric oxide.83,84 These studies were confirmed by independent research groups.71,85 This fundamental effect of inflammation induced inhibition of norepinephrine release reduces the sympathetic break on secretomotor neurones supporting neurogenic secretory diarrhoea (fig 4⇑). This has been described in Clostridium difficile toxin A induced intestinal inflammation and directly by the influence of IL-1β and IL-6.86,87 In addition, prostaglandins such as PGE1 and PGE2 released on inflammation attenuated the sympathetically induced inhibition of motoneurones.88 Furthermore, inhibition of norepinephrine release supports the chronicity of inflammation due to loss of sympathetic inhibition of innate and Th1 mediated immune responses (see above).
This phenomenon was not found in ulcerative colitis specimens,79,89 which supports the increase in sympathetic nerve fibres in this disease.78 With respect to sympathetic innervation, Crohn’s disease (and rheumatoid arthritis) seem to be remarkably different from ulcerative colitis. Thus it is likely that different immune or healing responses are used in these contrasting diseases.
Link between leucocyte infiltration, innervation, and motility disorders
We have focused on changes in the sympathetic nervous system in conjunction with intestinal inflammation. However, entities such as irritable bowel syndrome are also linked to inflammatory changes of the enteric nervous system. In a seminal paper, it was demonstrated that patients with irritable bowel syndrome demonstrated leucocyte infiltration of the myenteric plexus and neuronal degeneration.90 We do not know how leucocyte infiltration affects sympathetic pathways but, given the increased release of proinflammatory cytokines, one may expect a decrease in sympathetic neurotransmitter release.71,81,83–,87 Inflammation induced inhibition of norepinephrine release would reduce the sympathetic break on secretomotor neurones, supporting neurogenic secretory diarrhoea in irritable bowel syndrome (fig 4⇑). On the other hand, its has been demonstrated that patients with inflammatory bowel diseases demonstrate intestinal pseudo-obstruction and degenerative neuropathy in the myenteric plexus.91 As infiltrating leucocytes can also produce substance P or support cytokine driven substance P release, increased secretomotor tone might be the consequence of leading to obstructive symptoms. Thus it might well be that inhibition and stimulation of secretomotor neurones by infiltrating leucocytes may be present at the same time, leading to a heterogeneous picture of various symptoms. In addition, increased infiltration of enteroendocrine cells during inflammation may further complicate the situation because these cells produce neurotransmitters which can affect intestinal function.92
Adrenoceptor expression in gut inflammation: the β to α adrenergic shift
In chronic inflammation, reduced expression of β adrenoceptors and increased expression of α1 adrenoceptors were observed.93–,95 This shift was termed the β to α-adrenergic shift of adrenoceptor expression.96 Consequently, this shift would support a proinflammatory immune response (see above). A similar shift has not been described in patients with inflammatory bowel diseases. However, experimentally induced intestinal inflammation leads to a similar phenomenon: induction of colitis increased α2 adrenoceptor expression in both ileal and colonic muscular layers.85 Three days after intraperitoneal injection of zymosan, a decrease in β adrenergic smooth muscle contraction was observed.97 In the guinea pig, inflammation is accompanied by upregulation of α1 and α2 adrenoceptors but downregulation of β adrenergic receptors on smooth muscle membranes within 10 days of TNBS jejunitis.98 In addition, the increase in blood flow induced by the β adrenergic agonist isoproterenol was markedly lower in inflamed ileum.99 These studies in animal models of intestinal inflammation support the concept of a β to α adrenergic shift. The functional consequences are further corroborated by repulsion of sympathetic nerve fibres (fig 5B⇑).
We hypothesise that the β to α adrenergic shift has been evolutionarily conserved to overcome bacterial infection in the gut. Indeed, it has been demonstrated that α adrenergic signalling increases defence mechanisms in the gut.100 In addition, signalling through α2 adrenoceptors can be important for macrophages to resist the intracellular growth of microbes.58
Sympathetic nerve fibres in relation to substance P positive nerve fibres
It has been repeatedly demonstrated that substance P from the visceral nociceptive system and intrinsic sensory afferents is a proinflammatory mediator in intestinal inflammation.23,25 Similarly, this point of view is true for arthritis.101 Interestingly, in contrast with sympathetic nerve fibres, substance P positive nerve fibres sprout into inflamed tissue, as described in rheumatoid arthritis and inflammatory bowel diseases.25,65 As sympathetic neurotransmitters normally inhibit release of substance P, the preponderance of the sensory system over the sympathetic system is likely an additional inflammatory factor.
Sympathetic nervous tone in inflammatory bowel diseases
Similar to other chronic inflammatory diseases,102–,106 the tone of the sympathetic nervous system is increased in patients with inflammatory bowel diseases.107 In rheumatoid arthritis and systemic lupus erythematosus, this phenomenon was related to increased mortality.108,109 We mentioned above that norepinephrine can be anti-inflammatory at increased concentrations (targeting cells of the innate immune system). However, an elevated sympathetic nervous tone does not increase norepinephrine at the site of inflammation due to loss of sympathetic nerve fibres (see above). In addition, concentrations of circulating norepinephrine are only increased by a factor of 2–3 (from approximately 10−9 M to 2−3×10−9 M). Thus increased plasma concentration of this neurotransmitter is not sufficient to significantly increase the local concentration in inflamed tissue. On the other hand, the increased systemic concentration of norepinephrine might facilitate leucocyte mobilisation from non-inflamed areas, which may then migrate to sites of inflammation.
In common with rheumatoid arthritis,110,111 patients with inflammatory bowel diseases show inadequately low concentration of the anti-inflammatory steroid hormone cortisol in relation to inflammation, as measured by IL-6 and TNF.107,112 Consequently, this also leads to low concentrations of cortisol in inflamed tissue. Considering the above mentioned cooperativity of cortisol and norepinephrine, low cortisol concentrations in relation to inflammation is an additional chronicity factor.
CONCLUSIONS
Striking similarities exist between rheumatoid arthritis and inflammatory intestinal disease:
Rheumatoid arthritis and Crohn’s disease are disorders with a Th1 lymphocyte dominance, signs of an activated innate immune system in the chronic symptomatic phase (macrophage, neutrophils and others), and overshooting responses of myofibroblasts/fibroblasts leading to scar/pannus formation.
The inflammatory reactions described above are typically suppressed by the SNS via β adrenergic pathways. This happens directly at the level of T lymphocytes, macrophages, dendritic cells, NK cells, and neutrophils.
Loss of sympathetic nerve fibres in inflamed tissue and inflammation induced inhibition of norepinephrine release with a concomitant decrease in local neurotransmitter levels converts a normally present β adrenergic zone into an α adrenergic zone.
This is followed by a reduction in the sympathetic break on secretomotor neurones leading to secretory diarrhoea and by an overall proinflammatory microenvironment.
In addition, a preponderance of proinflammatory substance P positive nerve fibres in relation to sympathetic nerve fibres supports these phenomena.
The parallel inadequate secretion of cortisol together with loss of β adrenergic receptor mediated effects leads to inadequate anti-inflammatory cooperativity of these two systems.
All of the above mentioned factors contribute to an overall proinflammatory milieu at the local site. The similarities between the two diseases of rheumatoid arthritis and Crohn’s disease suggest that a general principle exists which can explain these facets in chronic inflammation. We recently hypothesised that the observed changes in these diseases have been evolutionarily conserved to overcome infectious diseases and to support the wound healing process.113 In chronic inflammatory diseases, these evolutionarily conserved mechanisms are also used but are probably inadequate. This review reinforces the belief that apart from the immune system, many other factors contribute to the chronic inflammatory process. As a consequence, more wider ranging and well adjusted combination therapy must be introduced, which comprises additional aspects together with the immune system (neuroendocrine targets) because the immune system is not a lonely player in an empty body.
Acknowledgments
This work was supported by the DFG (SFB 585, TP B8).
REFERENCES
Footnotes
Conflict of interest: None declared.