Article Text

Statistics from Altmetric.com
Alcoholic hepatitis (AH) is a severe form of alcoholic liver disease that is associated with high mortality. Despite extensive research on AH over the last several decades, the pathogenesis of AH remains largely unknown, and there are no approved targeted therapies. Inflammation (neutrophil infiltration) is generally believed to play an important role in the pathogenesis of AH; however, the mechanisms underlying neutrophil infiltration in AH and the functions of neutrophils in AH are not fully understood.1 Despite its obscure role, inflammation has been actively investigated as a therapeutic target for the treatment of AH. For example, corticosteroids, which are broadly immunosuppressive drugs, have been widely used for AH therapy for more than five decades. However, the results have been controversial. It is generally accepted that corticosteroid therapy improves short-term, but not long-term, survival rates in patients with severe AH.2
Recently, we have demonstrated that treatment with prednisolone, a synthetic steroid drug, reduced hepatotoxin (eg, ethanol and CCl4)-induced liver injury in mice by inhibiting neutrophil-mediated phagocytosis and liver regeneration, suggesting a detrimental effect of steroid therapy in hepatotoxin-induced hepatitis.3 Additionally, it is known that steroid therapy is ineffective and, occasionally, even detrimental for the treatment of neutrophil-mediated diseases because steroid treatment stimulates bone marrow neutrophil release and increases neutrophil survival.4 Based on this phenomenon, it is plausible to speculate that steroid therapy for AH may have no beneficial effects on the injured liver itself but may retain certain beneficial effects in attenuating systemic inflammatory responses and, thus, improving the short-term survival rate. Therefore, there is an urgent need to identify specific targets that can inhibit liver inflammation (neutrophil infiltration) for the treatment of AH.
Over the last three decades, studies using rodent models of chronic alcohol feeding identified many inflammatory mediators that play important roles in the pathogenesis of chronic alcoholic liver injury.1 However, most of these chronic ethanol-feeding models exhibit no or low levels of hepatic neutrophil infiltration. Therefore, many of the identified inflammatory mediators may not contribute to the neutrophil infiltration observed in acute AH. Recently, we developed a chronic-binge ethanol-feeding model that presents significant neutrophil infiltration.5 Studies using this model will likely aid in identifying several inflammatory mediators of neutrophil infiltration in AH. However, this chronic-binge ethanol-feeding model only represents some features of moderate AH, and currently there are no animal models that reproduce the full spectrum of human AH. To overcome the shortcomings of animal models, the Bataller group analysed human AH biopsy samples by real-time PCR and microarray. Their early studies using real-time PCR analyses revealed that the levels of several CXC subfamily members, including IL-8, Gro-α, CXCL5, CXCL6, CXCL10 and platelet factor 4, are significantly elevated in AH livers compared with normal healthy control livers, and are correlated with neutrophil infiltration and the severity of portal hypertension.6 The CC chemokine CCL2, but not CCL5, was also found to be upregulated. Higher expression levels of IL-8, CXCL5, Gro-γ, and CXCL6 were associated with a worse prognosis. Recently, the Bataller group performed microarray analyses of human AH samples, and further demonstrated that the levels of many CXC and CC family members are markedly elevated in AH biopsy samples compared with healthy control liver samples.7 Among these chemokines, CCL20 is one of the most upregulated chemokines in AH liver samples.7
CCL20, also called liver and activation-regulated chemokine (LARC), macrophage inflammatory protein-3α (MIP-3α), or Exodus-1, is expressed in several tissues, with the highest expression observed in peripheral blood lymphocytes, lymph nodes, the liver, the appendix and the fetal lung. CCL20 expression can be further induced by many inflammatory mediators, such as lipopolysaccharides (LPS), TNF-α, and IL-1β. The major function of CCL20 is to attract lymphocytes, and to a much lesser extent, attract neutrophils via the binding and activating of the chemokine receptor CCR6 on these cells. Thus, CCL20 plays an important role in regulating adaptive immunity and autoimmunity. In the liver, the interaction between CCL20 and CCR6 has been shown to regulate liver inflammation, fibrosis and tumorigenesis.8–10
Affo et al11 performed a high-quality translational study of the functions of CCL20 in AH by analysing human AH samples and using mouse models of liver injury under acute-on-chronic conditions. Their examination of human AH samples revealed that the hepatic and serum CCL20 levels are highly elevated in patients with AH and are correlated with the degree of fibrosis, portal hypertension, endotoxaemia, disease severity score, and short-term mortality. Furthermore, Affo et al used animal models of liver injury induced by CCl4 plus ethanol or CCl4 plus LPS and demonstrated that macrophages and hepatic stellate cells are the main CCL20-producing cell types (figure 1). Additionally, silencing CCL20 in vivo ameliorated LPS-induced liver injury and inflammation.
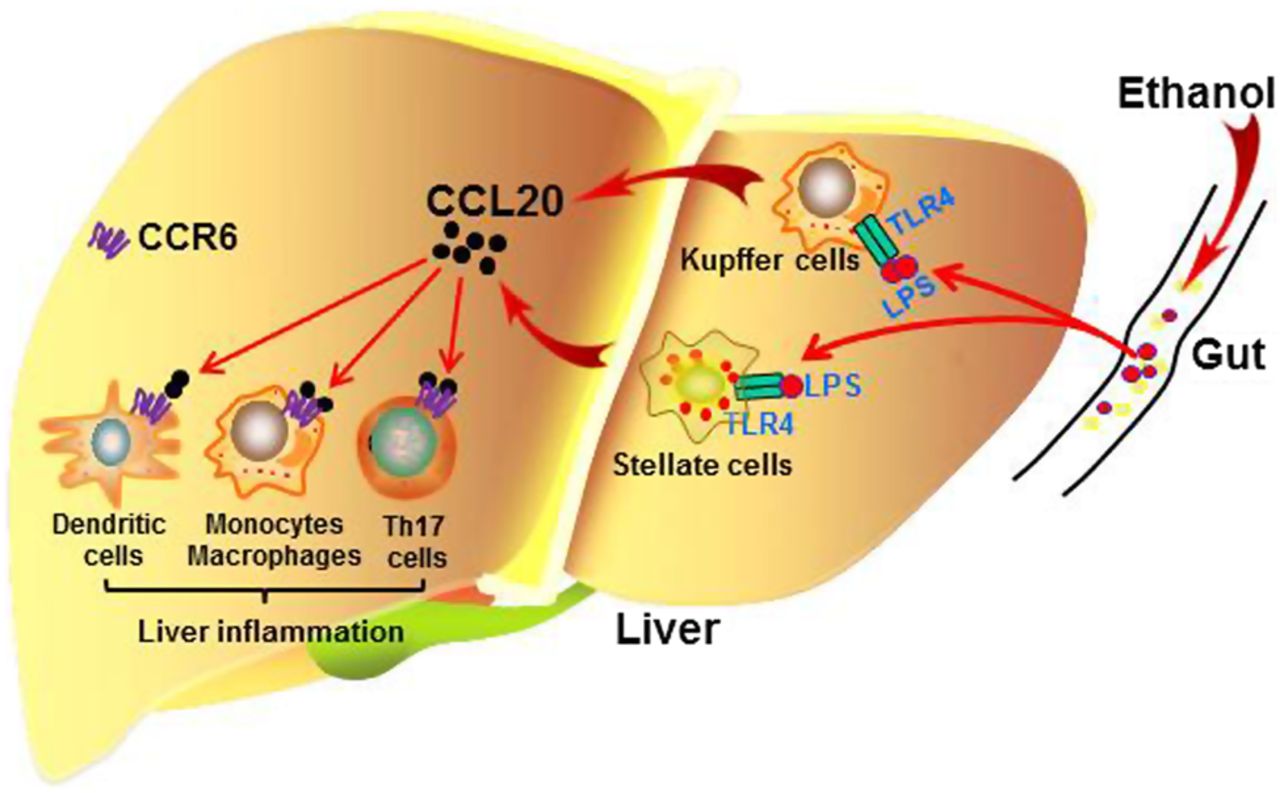
{kind=link}
A model depicting the roles of CCL20 in alcoholic steatohepatitis. Excessive alcohol drinking results in elevated levels of bacterial products, such as lipopolysaccharides (LPS), in the portal blood. LPS, via targeting TLR4, stimulates Kupffer cells and hepatic stellate cells to produce CCL20. This chemokine activates dendritic cells (DCs) and monocytes/macrophages to produce proinflammatory cytokines by targeting CCR6, leading to liver inflammation.
Although the authors demonstrated that the hepatic and serum CCL20 levels positively correlate with disease severity in AH patients, and that CCL20 plays an important role in LPS-induced liver injury, the mechanism by which CCL20 contributes to the pathogenesis of AH remains unknown. Because it strongly promotes the chemoattraction of DCs, Th17 cells and monocytes. but only weakly attracts neutrophils, CCL20 may directly activate and attract these lymphocytes. These cells then produce inflammatory mediators and chemokines that subsequently cause neutrophil infiltration in AH (figure 1).
Studies by Dr Bataller's group and other groups have demonstrated that a variety of chemokines are highly upregulated in AH and that many of these chemokines are correlated with disease severity. The obvious question is whether these chemokines can be therapeutically targeted for the treatment of AH. So far, many chemokine receptor antagonists have been developed, but most of these have failed in clinical trials for the treatment of several inflammatory diseases except for two approved drugs for non-inflammatory diseases: a CCR5 inhibitor, which is used to treat HIV, and a CXCR4 antagonist, which serves as a haematopoietic stem cell mobiliser.12 One of the most important reasons for these failures is target redundancy (one chemokine can target several receptors and vice versa) in complex inflammatory diseases. The pathophysiology of AH, especially the severe form, is very complex, with elevations in the levels of multiple chemokines, including IL-8, Gro-α, CXCL5, CXCL6, CXCL10, CCL2 and CCL20, among others.6 ,11 These chemokines likely work together either additively or synergistically to promote liver inflammation in AH patients by targeting a wide variety of chemokine receptors on various types of inflammatory cells, non-parenchymal cells and hepatocytes. There are no data or ways to demonstrate which chemokine plays an essential role in the pathogenesis of AH due to a lack of animal models that reproduce all the key histological features of human AH. Thus, it is reasonable to speculate that it will be very difficult to demonstrate clinical efficacy for the treatment of AH by targeting a specific antagonist to a single receptor because of chemokine receptor redundancy. The development of promiscuous chemokine receptor antagonists that can target several G-protein-coupled chemokine receptors may be required for the treatment of AH. Future translational studies that employ a combination of animal models and clinical AH samples are also urgently required to identify the chemokine(s) that critically contribute to liver inflammation in AH so that we can identify which chemokine(s) should be targeted for AH therapy.
References
Footnotes
-
Contributors Both authors contributed to writing the commentary and generating the figure and approved the final version.
-
Competing interests None.
-
Provenance and peer review Commissioned; internally peer reviewed.