Article Text

Statistics from Altmetric.com
Summary
Positional cloning and linkage analysis have shown that inactivation of one of the mismatch repair genes (hMLH1, hMSH2, hPMS1, hPMS2, GTBP/hMSH6) is responsible for the microsatellite instability or replication error (RER+) seen in more than 90% of hereditary non-polyposis colorectal cancers (HNPCC) and 15% of sporadic RER+ colorectal cancers. In HNPCC, a germline mutation (usually in hMLH1 or hMSH2) is accompanied by one further event (usually allelic loss) to inactivate a mismatch repair gene. In contrast, somatic mutations in the mismatch repair genes are not frequently found in sporadic RER+ colorectal cancers. Hypermethylation of the hMLH1 promoter region has recently been described, and this epigenetic change is the predominant cause of inactivation of mismatch repair genes in sporadic RER+ colorectal and other cancers. Inactivation of a mismatch repair gene may occur early (before inactivation of the APC gene) and produce a raised mutation rate in a proportion of HNPCC patients, and these cancers will follow a different pathway to other RER+ cancers. However, it is likely that selection for escape from apoptosis is the most important feature in the evolution of an RER+ cancer.
Historical background to hereditary non-polyposis colorectal cancer (HNPCC)
Long before molecular genetics had given us insight into the aetiology of colorectal cancer, Dr Aldred Warthin, professor of pathology at the University of Michigan, Ann Arbor, had described several families who appeared to have a predisposition to cancer.1 ,2 In 1895, Dr Warthin's seamstress had commented that she would die of gastric, colon, or uterine cancer. She stated that most of her relatives had died from these conditions and she did indeed die from endometrial carcinoma at a young age.
In 1966, Dr Henry Lynch from Omaha, Nebraska, and Dr Marjorie Shaw from Ann Arbor, Michigan, published the findings of two large families (family “N” from Nebraska and family “M” from Michigan) that had a large number of individuals with multiple primary cancers.3 The families were also of interest as there was a varied distribution of cancer, a high incidence of endometrial cancer, and the cancers were transmitted through several generations. This work prompted interest from Dr A James French who had succeeded Dr Aldred Warthin as chairman of pathology at the University of Michigan. Having read about families “N” and “M”, he gave custody of all the detailed records and pathology specimens that had been collected by Dr Warthin over 30 years to Dr Lynch. This work resulted in an updated review of cancer family “G” (the family of Dr Warthin's seamstress) and demonstrated an autosomal dominant pattern of inheritance, with the majority of cancers being adenocarcinomas of the colon, endometrium, and stomach.4 Although gastric cancers were the predominant cancers in the early generations of the family, this was replaced later by colorectal cancers. This mirrors the change in the incidence of sporadic gastric and colorectal cancers in the general population, and presumably reflects environmental influences.
Throughout the 1970s and early 1980s there remained a great deal of scepticism that cancer could have a strong hereditary component and the work of Warthin and Lynch was seen as being anecdotal. However, by the 1980s many reports of a “cancer family syndrome” were appearing in the medical literature.5 ,6 Cancer family syndrome then became subdivided into Lynch syndrome I (families with mainly colorectal cancers at an early age) and Lynch syndrome II (families with colorectal and extracolonic cancers, particularly of the female genital tract).7 All of this different terminology was eventually clarified with the introduction of the term hereditary non-polyposis colorectal cancer (HNPCC) to emphasise the lack of multiple colonic polyps and to separate it from the polyposis syndromes.
Discovery of human mismatch repair genes
Following the study of large kindreds using linkage analysis, the HNPCC susceptibility loci were mapped to chromosome 2p16 and chromosome 3p21.8 ,9 Expanded microsatellites were found in HNPCC rather than regions of loss as had been expected, and this was termed microsatellite instability.10 Microsatellite instability had already been studied extensively in bacteria and yeast and this led to positional cloning strategies identifying the human homologue for the mutS gene (hMSH2—humanMutS homologue) on chromosome 2p,11 ,12 followed closely by the identification of the human homologue of the mutL gene (hMLH1—humanMutL homologue) on chromosome 3p (table 1).13 ,14. Mutations in hMSH2 and hMLH1 account for the majority of reported HNPCC cases,15although two additional homologues of the mutL gene (hPMS1 on chromosome 2q and hPMS2 on chromosome 7q) have been cloned and mutations found in a small number of HNPCC kindreds.16 Two other homologues of the mutS gene have also been cloned (hMSH3 and GTBP/hMSH6)17 ,18 and mutations have recently been described in GTBP in HNPCC kindreds,19 ,20 with a somatic mutation previously reported in GTBP in a colorectal cancer cell line.21 These genes, and the proteins they encode for, are responsible for eukaryotic mismatch repair.
Bacterial mismatch repair enzymes and their human homologues
Microsatellite instability
Microsatellites are repetitive genetic loci (1–5 base pairs, repeated 15–30 times) which are normally relatively stable, and microsatellite instability (or replication error positive, RER+) is defined as a relatively frequent change of any length of these loci due to either insertion or deletion of repeated units. They are prone to slippage during DNA replication and this results in a small loop in either the template or nascent DNA strand. These are normally repaired, but in the absence of efficient mismatch repair function, these “loops” may become permanent, and alleles of different sizes will be formed at the next round of replication. Hundreds of thousands of microsatellites are present throughout the human genome and are susceptible to insertion/deletion mutations. When they are found in the introns of genes, they often result in polymorphic changes because mutation is relatively common even in normal cells. Most people then have two alleles with a different but constant number of repeated units found throughout their cells, and often a different pattern is seen between individuals as the microsatellite loci are particularly polymorphic.
Microsatellite instability is seen when new alleles are formed relatively frequently at microsatellite loci in tumour DNA compared with the two parental alleles in the normal DNA of the patient. If a cancer has a mutated mismatch repair gene, multiple different sized alleles may accumulate over several generations. While microsatellite instability is seen in about 90% of all HNPCC and at the majority of microsatellite loci,10 ,22 it is only seen in 10–15% of sporadic colorectal cancers.23 Microsatellite instability is also seen in cancer from other organs (although at a lower frequency), including endometrial cancer, ovarian cancer, pancreatic and gastric cancer, and keratocanthoma.24 The relative frequency of microsatellite instability is higher in pancreatic, endometrial, prostate, and gastric carcinomas (which are found commonly in Lynch II pedigrees) than in breast, ovarian, and other carcinomas.24
Disagreement between authors as to how precisely microsatellite instability should be defined has led to difficulties in comparing scientific work from different institutions. Criteria for defining the RER+ phenotype range from microsatellite instability observed at just one locus, to microsatellite instability at a proportion (for example, 30%) of loci studied.23 ,25 ,26 Some workers have tried to distinguish between tumours with many new alleles and those with only a few new alleles at these loci.27 More recently authors have described the use of BAT26, a poly (A) tract localised in the fifth intron of hMSH2, to define whether or not a tumour is RER+.28 This microsatellite has proved both highly sensitive and specific for this purpose, and has the additional advantage of not requiring constitutional DNA for comparison as in RER+ cells it produces abnormal patterns. The National Cancer Institute recently published criteria for the determination of microsatellite instability in colorectal cancer.29 Using a panel of five microsatellites, including BAT26, a cancer is described as having microsatellite instability if two or more of the five markers are unstable. This follows the “Bethesda guidelines” which were set up to assist in the decision of which colorectal cancers should be tested for microsatellite instability.30 This should help identify some HNPCC patients with germline mutations who do not fulfil the “Amsterdam criteria” (table 2).
Amsterdam criteria for hereditary non-polyposis colorectal cancer
Hereditary non-polyposis colorectal cancer (HNPCC)
HNPCC is an autosomal dominantly inherited disorder of cancer susceptibility with high penetrance (80–85%),31 and to date mutations have been described in five mismatch repair genes—hMSH2, hMLH1, hPMS1, hPMS2, and GTBP (MSH6).12-14 ,16 ,19 ,20 It has been previously estimated that HNPCC is responsible for 5–10% of all colorectal cancers32 although a prospective study from Finland has demonstrated that only 10 of 509 (2%) consecutive colorectal cancer patients had germline mutations in either hMLH1 or hMSH2.33 This may still be an overestimate as five of the 10 patients had the Finnish founder mutation 1.
Traditionally, the HNPCC syndrome has been divided into Lynch syndromes I and II although with the progress of molecular genetics it is accepted that these divisions are artificial and do not represent two diseases with mutations at separate genetic loci. Lynch syndrome I patients tend to have carcinoma of the right colon (70% of colorectal cancers proximal to the splenic flexure) with many patients presenting with synchronous and metachronous tumours.31 ,34 Lynch syndrome II patients have similar colorectal cancer pathology but in addition have extracolonic cancers. These include carcinomas of the endometrium, ovary, stomach, pancreas, small bowel, hepatobiliary tract, and the ureter and renal pelvis.35 Sebaceous adenomas and carcinomas, and keratocanthomas present together with other features of HNPCC make up the Muir-Torre syndrome,36while glioblastomas (and multiple colonic adenomas) present in Turcot's syndrome.37 Muir-Torre syndrome with its skin lesions, and Turcot's with its colonic adenomas, possess the only clinical markers of HNPCC kindreds before the onset of cancer, although recently microsatellite instability has been demonstrated in benign skin lesions of HNPCC kindred who have not yet developed cancer.38
The majority (70%) of HNPCC patients have a germline mutation in either hMSH2 or hMLH,39 giving a lifetime risk of about 80% for colorectal cancer.40 ,41 The results of a large collaborative study showed that 83% of hMSH2 germline mutations in HNPCC patients were either nonsense or frameshift mutations, and this contrasts with 49% of hMLH1 germline mutations.42 Thirty one percent of hMLH1 germline mutations are missense changes. The mutations were evenly distributed across both the mismatch repair genes.
Males are at greater risk (to the age of 70) of developing a cancer (91% v 69%) and the risk of developing colorectal cancer is also significantly higher in males than females (74% v 30%).40 In females the risk of developing an endometrial cancer is higher than the risk of developing a colorectal cancer (42% v30%).40 It has been suggested that the risk of endometrial carcinoma is greater in those patients who have a mutation in hMSH2 rather than hMLH1 (61% v42%).41
Clinical selection criteria for families with HNPCC were established by the International Collaborative Group on HNPCC (ICG-HNPCC) in Amsterdam in 1990 (table 2).43 These were produced to provide uniformity between researchers and in collaborative studies. However, the criteria have been criticised as they exclude extracolonic cancers that may be present in a classic HNPCC family. The criteria may thus prevent the diagnosis of HNPCC in small families and conversely lead to a false diagnosis of HNPCC in large families purely by the chance clustering of cancers. Revised criteria have recently been published that take into account the common occurrence of extracolonic cancers in HNPCC (cancer of the endometrium, small bowel, ureter, or renal pelvis).44
Sporadic colorectal cancer and microsatellite instability
Microsatellite instability is seen in nearly all HNPCC but only in 10–15% of sporadic colorectal cancers.23 ,45 Both HNPCC and sporadic RER+ cancers are diploid or near diploid in chromosomal constitution. This is in direct contrast with the majority of colorectal cancers (and most carcinomas in general) which are aneuploid in chromosomal constitution.10 ,22 ,25
Microsatellite instability is a result of inactivation of both alleles of a mismatch repair gene, in accordance with Knudson's hypothesis for tumour suppressor genes.46 In HNPCC, a single mutation is inherited in the germline and microsatellite instability only follows inactivation of the other allele, otherwise the mutation rate is normal. In sporadic RER+ cancers, inactivation of both alleles must occur somatically before microsatellite instability is observed.47 ,48
Although germline mutations in hMLH1 and hMSH2 are commonly found in HNPCC,39 somatic mutations have not been described frequently in presumed sporadic colorectal cancers.48Somatic mutations in hMLH1 and hMSH2 have been described only rarely in other solid cancers, such as endometrial and ovarian, with microsatellite instability.49 ,50
The lack of detectable mutations in hMLH1 and hMSH2 in sporadic cancers with microsatellite instability led to the hypothesis that there may be other genetic loci for encoding proteins responsible for DNA mismatch repair. Thibodeau et al demonstrated that 40 of 42 (95%) sporadic RER+ colorectal cancers lacked expression of either hMLH1 or hMSH2,51 and that hMLH1 was the altered protein in 95% of these cases. They concluded that hMLH1 has a principal role in the phenotype of sporadic RER+ colorectal cancer.
DNA mismatch repair
Mispaired bases in eukaryotic DNA are first recognised by two heterodimeric complexes of MutS related proteins—MSH2/ GTBP (MutSα) and MSH2/ MSH3 (MutSβ) (fig 1).52-54 While it is thought that MutSα is responsible for base:base mispairs, MutSβ plays a major role in the repair of larger insertion/deletion mispairs.52 ,55 It is likely that both MutSα and MutSβ are unable to repair single base insertion/deletion mispairs. More recently, it has been shown that MSH2/GTBP complexes may support both the repair of base:base mispairs and insertion/deletion mispairs with up to 12 unpaired bases.56 Generally, MSH2/GTBP is best able to recognise and bind to a G:T mispair and +1 insertion/deletion loop mispairs, while MSH2/ MSH3 is best able to combine with +1 and larger insertion/deletion loop mispairs.18 ,57-60

Mismatch repair. A mispaired base is recognised by the hMSH2/GTBP complex while an insertion/deletion loop is recognised by the hMSH2/hMSH3 complex. MutL related proteins (hMLH1/hPMS2 and hMLH1/hPMS1 complexes) then interact with the MutS related proteins that are already bound to the mispaired bases. (The hMSH2/GTBP complex may also support the repair of insertion/deletion loops).
Following recognition of a mispair, a heterodimeric complex of MutL related proteins (MLH1/PMS1 (PMS2 in humans)) interacts with the MutS related proteins that are already bound to mispaired bases.61 ,62 MLH1/PMS1 binds to a MSH2/MSH3 mispair complex converting it into a higher molecular weight structure.63 MLH1 also forms a complex with MLH3 (PMS2 in humans) which plays a periodic role in the repair of insertion/deletion mispairs in the MSH2/MSH3 pathway.64 The MLH1/PMS1 complex increases the efficiency of MutS related proteins to recognise a mismatch.63 ,65
There are a number of other proteins involved in mismatch repair and these include DNA polymerase δ, replication protein A (RPA), proliferating cell nuclear antigen (PCNA), replication factor C (RFC), exonuclease 1, FEN1 (RAD27), and DNA polymerase δ and ε associated exonucleases.66
Once the DNA mismatch is recognised and the MutS and MutL heterodimer complexes have combined with it, repair of the mismatched DNA proceeds by activating exonulcease mediated degradation of DNA from a “nick” that is a distance of up to 1–2 kilobases from the mismatch.67 Degradation continues until the mismatched base is removed. The resulting long excision tract is filled in by DNA polymerase δ which inserts the correct nucleotide into the sequence.
Epigenetics and microsatellite instability
The majority of RER+ cancers have no detectable mutation in the mismatch repair genes. This led to speculation that novel genes were responsible or that non-mutational and epigenetic mechanisms resulted in microsatellite instability.47 ,48 ,68
Inactivation of several tumour suppressor genes by an epigenetic process that involves hypermethylation of the promoter region has been described.69-71 This results in transcriptional loss and subsequent lack of protein expression. The first links between microsatellite instability and methylation of promoter DNA followed the discovery that both endogenous and exogenous DNA sequences were more likely to be methylated in sporadic RER+ colorectal cancers.72 ,73 This finding together with so few somatic mutations being described in sporadic RER+ colorectal cancers led to the discovery of hypermethylation of the promoter region of hMLH1 being associated with lack of hMLH1 protein expression and microsatellite instability.74-78 Hypermethylation may be reversed in vitro, with subsequent re-expression of protein,78 and this raises the possibility of clinical applications. Subsequently, hypermethylation of the hMLH1 promoter has been described in up to 77% of sporadic RER+ endometrial carcinomas79 and in up to 100% of sporadic RER+ gastric carcinomas.80
The effects of gene expression that are not due to DNA sequence changes are referred to as epigenetic. It is thought that DNA methylation inhibits the initiation of transcription by reducing the binding affinity of transcription factors.81 ,82 Methylation occurs at the cytosine residues of CpG islands (GC and CpG rich areas) in the proximity of the promoter region of many genes.83Very little is known about the mechanisms responsible for methylation in normal or cancer cells, and some have suggested that abnormal methylation of DNA represents a “methylator” phenotype.83 Mammalian methyl transferases have recently been described,84 and it may be that a change in some aspect of the chromatin structure, possibly associated with histone acetylation,85 allows access for the these enzymes to the DNA.
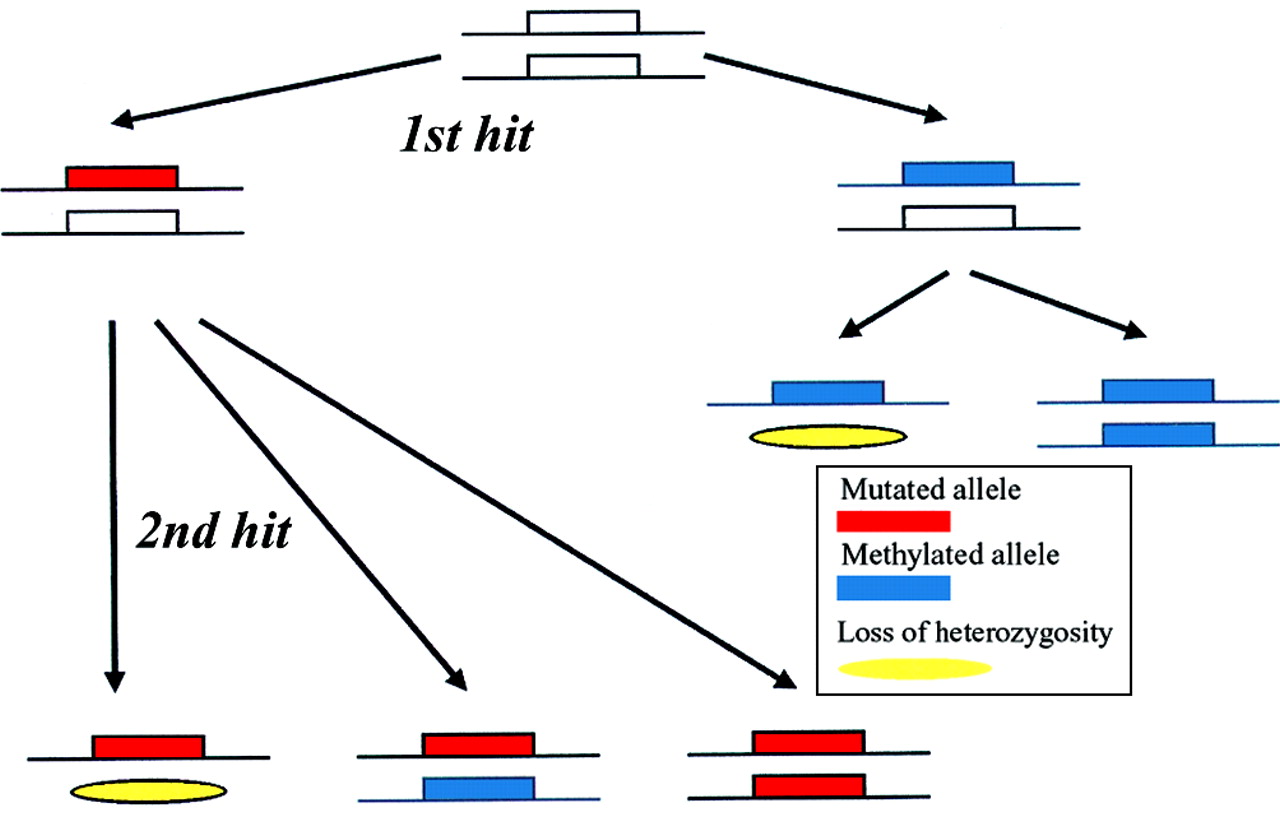
Knudson's hypothesis is fully accepted for the inactivation of tumour suppressor genes in cancer,46 and although it has traditionally focused on mutations in the DNA sequence and on loss of heterozygosity, it has been proposed that the hypothesis should be expanded to include epigenetic mechanisms of gene inactivation, such as methylation of the promoter (fig2).83 ,86.

Methylation and Knudson's two hit hypothesis. It has been proposed that epigenetic mechanisms, such as hypermethylation of the promoter region, should be included in the two hit hypothesis for inactivation of tumour suppressor genes. It was suggested that the first hit may be a mutation in the DNA sequence or promoter methylation. The second inactivating hit may be either loss of heterozygosity or a further mutational or methylating event in the second allele.
Mismatch repair genes and the “the adenoma-carcinoma sequence”
It has been proposed that HNPCC and sporadic RER+ colorectal cancers develop along a different pathway to other colorectal cancers, and these cancers are said to have an increased mutation rate or a “mutator phenotype” as a result of inactivation of a mismatch repair gene.87
Some HNPCC cancers may indeed have an accelerated pathway to carcinoma when loss of the remaining wild-type allele occurs early and before inactivation of the APC gene (fig 3). Although an excess of frameshift mutations in the APC gene have been reported in HNPCC and sporadic RER+ colorectal cancers compared with RER− cancers, this study did not apparently exclude familial cases.88 More recently, it has been shown that there is no difference in allelic loss at the APC loci, frameshift mutations in APC, or APC mutations in simple repeat sequences between RER+ and RER− sporadic (clearly non-familial) colorectal cancers.89 This suggests that APC mutations remain the initiating event in sporadic RER+ colon cancers although there may be an increased mutation rate in a proportion of HNPCC cancers in whom the mismatch repair gene is inactivated prior to APC mutations.

{kind=link}
{kind=link}
{kind=link}
Model for adenoma→carcinoma pathway in HNPCC versus sporadic RER+ colorectal cancers. In HNPCC, a germline mutation is present in every cell, and only one further event (usually loss of heterozygosity (LOH)) is required to inactivate a mismatch repair gene. This may occur at an early stage (A), before inactivation of APC, and result in rapid progression through the adenoma→carcinoma pathway. In contrast, inactivation of a mismatch repair gene in sporadic RER+ cancers is likely to be a late event, after inactivation of APC. Although inactivation may be due to a somatic mutation (with LOH), hypermethylation of the hMLH1 promoter region is the commonest cause of inactivation of mismatch repair genes in these cancers, and is usually a biallelic event (B, C).
In sporadic RER+ colorectal cancers, microsatellite instability may be a late event in the adenoma to carcinoma sequence, associated with a selective advantage of mismatch repair gene mutations not directly related to the increased mutation rate. The sporadic cancer may then have acquired a mutator phenotype as a “bystander” effect at a stage when the mutation rate is less limiting because of the large population size of tumour cells. There is experimental evidence to support this suggestion.90 It has also been shown that a mutator phenotype is not necessary for the outgrowth of colorectal cancer91 and this may explain the low frequency of microsatellite instability seen in sporadic colorectal cancer.
Selection for inactivation of a mismatch repair gene may be for resistance to apoptosis, as is the case for p53 mutations,92 and not an increased mutator phenotype.93 Recently, it has been shown that apoptosis can be induced by overexpression of hMSH2 or hMLH1,94 and this supports the hypothesis that HNPCC and sporadic RER+ cancers lose the ability to undergo efficient apoptosis, as previously suggested.93
The overall frequency of both APC and K-ras mutations has been demonstrated to be similar in sporadic colorectal cancers and HNPCC.10 RER+ colorectal cancers have a diploid or near diploid karyotype which is in contrast with RER− colorectal cancers.87 Even those RER+ colorectal cancers that have p53 mutations, which are associated with aneuploidy, mostly have a near diploid karyotype (Eshleman and colleagues95 and unpublished observations). The low frequency of allelic loss in cancers with microsatellite instability is a reflection of this diploid karyotype.23 HNPCC cancers presenting at an advanced stage have a relatively good prognosis, and this may be as a result of the lack of gross aneuploidy seen in common sporadic colorectal cancers,96 or due to mutations in β2microglobulin (a HLA associated protein) which probably allows selection for escape from immune responses to the many bystander mutations that result from the replication error phenotype.97 The resulting lack of β2microglobulin protects a tumour from direct cytotoxic T cell attack.98
Although p53 mutations cause chromosomal instability and aneuploidy in the majority of colorectal cancers there is a subgroup of tumours that have neither p53 nor mismatch repair gene mutations. Mutation in genes which control the relationship between mitotic cell division and chromosome segregation have recently been described in some colorectal tumours99 and this may explain the chromosomal instability seen in some colorectal cancers.
Once a colorectal cancer becomes MMR deficient, it is not just microsatellites that are at risk of insertion/deletion mutations. All nucleotide repeat sequences, including those in the coding region of the genome, are at risk of mutation. Inactivating mutations have been found in mono- or dinucleotide repeat sequences in a number of RER+ colorectal cancers at a higher frequency than in RER− cancers, and include genes which encode for the type II TGF-β receptor, the IGF-II receptor, and the Bax protein.100-102 However, because of the very high mutation rate at the repetitive sequences in these genes in RER+ cells, the functional significance of these changes is not always clear.
Pathology of HNPCC colorectal cancers
Both HNPCC and sporadic RER+ colorectal cancers have clinicopathological features that make them distinct from the more common sporadic colorectal cancers. The cancers are predominantly found in the right colon and present at a younger age. The pathology of these cancers differs from the common sporadic colorectal cancers by virtue of an increased number of mucinous and undifferentiated cancers.26 ,103 The undifferentiated cancers are associated with an increased lymphocytic infiltrate,104 ,105 which may be connected with the immune response to multiple mutant proteins. The combination of mucinous appearance with undifferentiation should lead the clinician to suspect microsatellite instability.106
Although colorectal cancers present at a younger age in HNPCC, adenomatous polyps are not found at an increased frequency in patients with a germline mutation in one of the mismatch repair genes.107 Adenomas in HNPCC patients tend to be large, villous, severely dysplastic, and occur more commonly in the proximal colon compared with adenomas in the general population.108These adenomas may also show microsatellite instability and have mutations in the coding regions of susceptible genes that have been associated with colorectal cancers showing microsatellite instability.22 ,109 Mutations in the APC gene occur in the polyps of HNPCC patients,109 and this confirms that the polyps can be sporadic initially.
Conclusion
The discovery of mismatch repair genes is a good model for the use of linkage analysis and positional cloning of a putative tumour suppressor gene. Through this it has been shown that inactivation of the mismatch repair genes is responsible for the RER+ phenotype in HNPCC and sporadic RER+ colorectal cancers. Although cancers in a proportion of HNPCC patients will have a raised mutation rate and follow a different pathway to other RER+ cancers, it is likely that selection for escape from apoptosis is the most important feature in the evolution of an RER+ cancer. Future work may well focus on the cause, and perhaps prevention and reversal, of the recently described epigenetic changes—that is, hypermethylation of the hMLH1 promoter region—that result in the RER+ phenotype.
Abbreviations used in this paper
- RER+
- replication error positive
- HNPCC
- hereditary non-polyposis colorectal cancer
- RPA
- replication protein A
- PCNA
- proliferating cell nuclear antigen
- RFC
- replication factor C