Article Text

Statistics from Altmetric.com
Ever since Gajdusek implicated a “slow virus” in the transmission of Kuru, the oral route of “infection” of spongiform encephalopathies has attracted considerable interest. Transmission studies in transmissible spongiform encephalopathies (TSEs) have currently become an even more important area of study due to the possible transmission of bovine spongiform encephalopathy (BSE) to humans resulting in “new variant” Creutzfeldt-Jakob disease (vCJD). Prusiner's hypothesis that TSEs are caused by small proteinaceous infectious particles “prions” took considerable time to gain widespread recognition among the scientific establishment. The importance of these discoveries was vindicated by awards of Nobel prizes to Gajdusek and more recently to Prusiner.
Two forms of prion proteins are recognised—normal host encoded cellular prion protein (PrPc) and its pathological malfolded isoform (PrPSc)—which accumulate in the brain in TSEs including vCJD. PrPc is a copper binding glycoprotein attached to the plasma membrane through a glycosyl-phosphatidyl-inositol anchor.1 ,2 Although both isoforms are identical in amino acid sequence, they differ in their secondary structure: PrPSc is high in β sheet content in contrast with the predominantly α helical structure of PrPc. Partial resistance to proteolytic degradation, detergent insolubility, and slower turnover rate also characterise the pathological isoform.1
Prion hypothesis
The post-translational conversion of PrPc to PrPSc represents the central event in TSE pathogenesis. According to the prion hypothesis, this conversion is promoted by the homotypic interaction between native prion protein and de novo generated or incoming PrPSc. The latter was equated to the TSE infectious agent.1 The concept of prions as infectious agents, which are devoid of nucleic acids and encipher strain specific properties in the tertiary structure of the protein, has its strengths and weaknesses.1 ,3 ,4 Alternative hypotheses hold that PrP serves as a packaging substance and/or receptor for the infectious agent, which includes an as yet undiscovered nucleic acid. Some imperfection of the prion hypothesis is revealed by the demonstration that TSE can be transmitted to mice without the formation of detectable PrPSc.5 Furthermore, PrPSc like proteins produced in vitro have not yet been shown to be infectious.6 The strongest evidence for the prion hypothesis comes from experiments with transgenic mice7showing that mice lacking the PrP gene are not able to propagate infectivity and develop the disease. Use of these mice in neural graft experiments has proved that PrPc is necessary for the spread of the TSE infectious agent along neuronal pathways.8 It was also established that expression of PrPc in peripheral tissues is an absolute requirement for the spread of prions from peripheral sites to the central nervous system (CNS).9
Prion proteins and the intestine: peripheral pathogenesis of TSEs
In spite of oral exposure being a likely transmission route not only for vCJD but also for other TSEs, many of the published studies use inoculation of the TSE infected brain homogenates directly into the brain of experimental animals. The obvious rationale is that in comparison with intracerebral inoculation, oral exposure to the TSE infected material is relatively inefficient and commonly results in longer incubation periods.10 ,11 These difficulties in oral transmission of TSEs not only favour a certain experimental strategy but also raise a number of questions related to the exact sequence of events which starts with ingestion of the infectious material and either leads to a fatal brain disease or causes no obvious problems.
POTENTIAL SITES OF ENTRY
Early presence of infectivity12 and PrPSc(see Beekes and McBride13) in the aggregated intestinal lymphoid follicles (Peyer's patches) after oral exposure to the TSE infectious material has indicated the most probable site for the intestinal uptake of prions. The follicle associated epithelium (FAE) of Peyer's patches is specifically equipped to allow transit of macromolecules, particles, and microorganisms from the intestinal lumen to the underlying lymphoid tissue. This unique function of the FAE is attributed to the endocytic activity of membranous (M) cells (comprehensively reviewed eleswhere14-16). M cells differ from absorptive enterocytes in their lack of closely packed microvilli, thick glycocalyx, and hydrolytic enzymes. These features make the apical membrane endocytic domains readily accessible in M cells. M cells take up luminal material by a variety of different mechanisms, which include phagocytosis (bacteria, large particles), fluid phase endocytosis (non-adherent material), and absorptive endocytosis via clathrin coated pits (adherent material, viruses). The uptake rate depends significantly on size and biochemical properties of the particles. It is important to note that expression of apical surface glycoconjugates and related ability of M cells to “sample” specific pathogens vary depending on species and location in the gut. Whether these regional and species differences apply to uptake of prions requires direct investigation.
According to filtration experiments performed almost 30 years ago, the scrapie infectious particles are excluded by 35 nm filters.17 Previous studies using ionising radiation suggested an even smaller size of the scrapie agent.18Considering that scrapie is an animal form of TSE, it is possible to speculate that BSE prions are of a similar size. It is not known if infectious particles of a different size are required to cause the disease when administered by oral compared with intracerebral routes. Although pathological prion proteins are usually found in multimeric fibrillar aggregates in the TSE brain, it has not been ruled out that food preparation and subsequent processing during digestion may release a monomeric form of PrPSc.
Present controversy about the exact size and physical makeup of the TSE infectious particles creates difficulties in understanding the likely mechanisms and rate of uptake of the TSE agent by M cells. However, it does not exclude the possibility that M cells are the portal of entry for prions. Moreover, early accumulation of PrPSc in the FAE in hamsters19 and lambs20 orally challenged with scrapie infected material reinforces the potential role of the FAE in the intestinal uptake of prions. In addition, data on PrPSc accumulation in tonsils21 and appendix22 suggests that M cells found in these areas23 ,24 may provide entry sites for prions. Detection of PrPSc in villus epithelium,25 where no M cells have ever been reported, indicates that M cell independent uptake of prions is also possible.
A recent study of cattle and sheep colonic permeabilities to PrPSc (see McKie and colleagues26) has highlighted the significance of these M cell independent pathways for TSE infectious agent translocation across the intestinal epithelium. This study demonstrates that in vitro colonic permeability to PrPSc is higher in cattle than in sheep. The authors suggest that bovine colonic mucosa allows easier access for macromolecules via a paracellular route. Albeit in vitro, these reported differences in permeability still indicate that susceptibility of different species to TSE may in part depend on their ability to accumulate prions following the oral challenge.
Passive paracellular transit of particles and macromolecules across the tight junctions, which provide continuity of the intestinal epithelial barrier, is generally accepted to be very limited in health. An active transcellular route may represent an alternative M cell independent pathway and could be more relevant to translocation of prions across normal epithelium. Columnar enterocytes can endocytose small amounts of intact proteins.15 Binding to specific receptors on the apical surface of intestinal epithelial cells can facilitate uptake of macromolecules27 and even mediate the transcellular transfer of bacteria.28
We have described expression of the 37/67 kDa laminin receptor (LR) in the human small intestinal mucosa of patients without overt neuropathology.29 This receptor binds PrP (see Rieger and colleagues30) and its presence at the luminal surface of the intestinal epithelium in approximately 40% of patients examined may represent a risk factor for oral TSE susceptibility. Although we are not yet able to confirm that LR present at the apical surface of enterocytes could be internalised after ligand binding, this receptor has been shown to internalise laminin coated gold particles in a melanoma cell line31 and to mediate virus entry into cultured hamster cells.32 It is intriguing that LR acts as a receptor for viruses (Sindbis virus32 and tick borne encephalitis virus33) which can be transmitted orally to cause encephalitis in humans.
There are other candidate molecules for the role of a receptor for prion proteins. Recent data obtained in a cell free system support the idea that PrPc itself could serve as a ligand and/or receptor for PrPSc (see Horiuchi and Caughey34). Normal host PrP has been reported to be expressed in the epithelium of the gastrointestinal tract.35-38 Although there is inconsistency in the results between different research groups, particularly regarding the exact cellular location of PrPc, the potential role of this molecule in the epithelial uptake of the TSE infectious agent requires further study.
It is well known that increased permeability to macromolecules represents a normal feature of neonatal gut epithelium. The decreased number of Peyer's patches in older animals39 may also contribute to their reduced susceptibility to oral TSE infection. In humans, the number of Peyer's patches was reported to be higher in the intestines of teenagers than in young children or adults.40 Assuming that particle uptake capacity is a function of the area occupied by the epithelium overlying Peyer's patches, these differences may explain susceptibility of young people to vCJD.
Intestinal permeability and nutrient uptake are subject to genetic regulation41 ,42 and similar genetic control may be of relevance to the uptake of prion proteins. Although the latter is only speculative, one should consider its possible interaction with known genetic factors affecting susceptibility to TSEs, namely host PrP genotype.1 ,7 ,43 There is also a possibility that increased intestinal permeability44 associated with infection or inflammation may facilitate oral TSE infection.
Investigation of the exact portals of entry for the TSE infectious agent is important for a better understanding of the transmission of the disease. However, it may also help to resolve one fundamental problem of TSE pathogenesis, namely the apparent lack of immune response to the TSE agent. It is often argued that prions do not induce an immune response because they are “self proteins”. However, this is only partially true. When PrPSc, an important or even the only constituent of the TSE agent, enters a host belonging to a species different from the donor of the infectious tissue, it does represent “foreign material”, a potential antigen. It is only when the host organism starts to produce and accumulate its own PrPSc (as a result of conformational modifications of the host encoded PrPc) that this newly synthesised PrPSc may be regarded as a “self protein”.
There is a view that antigen uptake by M cells commonly results in an active mucosal and systemic immune response, probably due to the quick transport of antigens to specialised antigen presenting cells and subsequent expansion of helper T cells. How to reconcile the potential role of M cells in the uptake of prions with an apparent lack of an immune response remains unclear. On the other hand, villus enterocytes have been implicated in antigen presentation leading to induction of suppressor T cell populations and oral tolerance. Even if the real situation is more complex and influenced by many factors,27 it seems that the very possibility of an early and transient state of oral tolerance or local immune response to prions is worthy of investigation.
INITIAL ACCUMULATION OF PrPSc: PROSPECTS FOR EARLY DIAGNOSIS AND INTERVENTION IN ORALLY TRANSMITTED TSEs
Although some studies indicate that TSE can be transmitted without the formation of detectable PrPSc (see Lasmezas and colleagues5) and PrPSc is not necessarily associated with infectivity,45 this protease resistant form of the prion protein has been widely accepted as a marker of infectivity and used both for diagnostic purposes and as a “tracking device” to investigate pathways of the TSE infectious agent spread within the body.
Localisation of the PrPSc in Peyer's patches and other lymphoid organs in animals infected with scrapie has indicated follicular dendritic cells (FDC) as important elements in the propagation of the infectious agent.46 The exceptional ability of FDC to support replication of the TSE agent in lymphoid tissues47 has been used recently to develop a promising intervention strategy.48 A significant delay in the development of the experimental TSE has been achieved by temporary inactivation of FDC but as the authors used intraperitoneal inoculation of the TSE agent, the efficacy of FDC inactivation in delaying TSE development has yet to be proved for oral transmission. Nevertheless, this study clearly indicates a therapeutic potential for targeting specific cell populations which accumulate infectivity and PrPSc early in the development of peripherally transmitted TSE diseases.
Studies in rodents13 and sheep20 have demonstrated that after oral exposure to TSEs, PrPScaccumulates in gut lymphoid tissues prior to its appearance in the central nervous system (CNS), reinforcing the role of these tissues in the initial accumulation of PrPSc. Accumulation of PrPSc has also been reported in Peyer's patches and tonsils in non-human primates after natural and experimental oral infection by BSE agents.25
As PrPSc has been found in lymphoid tissues21 ,22 of people with vCJD, a sensitive immunohistochemical technique has been used to detect PrPScin a retrospective series of over 3000 tonsillar and appendiceal specimens.49 No positives have been detected so far but further larger studies are required. The ethical issues of conducting prospective studies to screen tissues derived from the intestine and its appendages for an incurable fatal disease are currently being examined very intensively. There is controversy on the reliability of current vCJD screening tests.50 Hence the size of the current “epidemic” of vCJD remains uncertain. The number of tonsils investigated in patients with established vCJD is small. Consequently, the presence of PrPSc in these peripheral tissues in all patients infected with vCJD is hard to estimate with certainty. This uncertainty is also fuelled by data describing the development of TSE without the formation of detectable PrPSc in cross species transmission experiments.5 Moreover, some breeds of sheep, naturally exposed to scrapie, develop disease but show no detectable infectivity in peripheral tissues.51 Although infectivity has been detected in the terminal ileum of cattle experimentally exposed to a high dose of the BSE agent,52 the presence of infectivity and/or PrPSc has not been reported in bovine intestines in field cases of BSE. In addition, after the initial accumulation of PrPSc in Peyer's patches, following experimental oral exposure to TSE, there is often a period when PrPSc is undetectable in Peyer's patches, even when a very sensitive immunohistochemical procedure is applied.20Perhaps we need an additional diagnostic test, which would not solely rely on detection of PrPSc but use some alternative marker.
Accumulation of PrPSc has been reported in the intestinal epithelium19 ,20 ,25 and in the ganglia of the enteric nervous system (ENS)19 ,20 in animals orally exposed to the TSE agent. The diagnostic and therapeutic potential of these findings has yet to be fully addressed and the presence of PrPSc in these locations in humans with vCJD has still to be investigated.
POTENTIAL PATHWAYS TO THE CNS
There is debate concerning haematogenous and neural pathways of the spread of TSE infectivity from the periphery to the CNS. The spread of TSE agent from the gastrointestinal tract might differ from such in other modes of peripheral transmission.12 Oral exposure to TSE agents results in slower spread of infectivity not only in comparison with intracerebral but also intraperitoneal inoculations.53 In contrast with the intraperitoneal challenge, the onset of clinical disease was not delayed by splenectomy performed prior to the experimental oral infection,12indicating that the lymphoreticular system and haematogenous spread of the infectivity may play a secondary role in orally transmitted TSEs. Data showing early accumulation of PrPSc in the ENS of hamsters orally infected with scrapie provided new evidence for an important role of the ENS in TSE neuroinvasion.19 There is still considerable difficulty in experimentally separating the roles played by neuronal and haematogenous pathways. The breakthrough seems to be recently achieved by a study of oral and intraperitoneal infection with hamster scrapie in transgenic and “knockout” mice which express hamster PrPc in nerves under the control of neurone specific enolase.54 The authors demonstrated that some strains of TSE agent may bypass the lymphoreticular system and proceed directly to the brain via peripheral nerves.
Direct investigation of TSE neuronal spread in humans is not possible due to obvious ethical limitations. However, the fact that expression of normal host prion protein (PrPc) is required for the spread of infectivity from peripheral sites to the CNS9provides an opportunity to elucidate potential pathways of TSE spread by localising specific neuronal elements which express PrPc. Recently, in collaboration with our colleagues from the National CJD Surveillance Unit (Edinburgh) and Institute for Animal Health (Edinburgh), we have presented the first evidence that PrPc is constitutively expressed in the human ENS.55
We described the anatomical and cellular distribution of PrP gene and protein in enteric ganglia and neuronal elements of the mucosa in histologically normal small intestinal biopsies/surgical resection samples from patients with no clinically manifest neuropathology. These data have been corroborated by investigation of PrPcexpression in the ENS of wild-type (Prnp+/+) and PrP knockout (heterozygous Prnp0/+ and homozygous Prnp0/0) mice. In situ hybridisation revealed PrP mRNA both in nerve cell bodies and in glial cells within the ENS ganglia. Intimate association of PrPc positive nerve endings with epithelial cells presented a characteristic feature of normal prion protein expression in all samples examined (fig 1). This implies that only a thin layer of intestinal epithelium stands between ingested TSE agent and host PrPc in the enteric nerves. Following oral TSE challenge, one of the early targets may therefore be the human ENS, providing not only the route of spread but also a site for the initial generation of infectivity and PrPSc. The presence of the potential receptor for prion proteins at the luminal surface of the intestinal epithelium29 may enable transfer of the infectious agent across the human gut epithelium to the adjacent PrPc positive nerve endings.
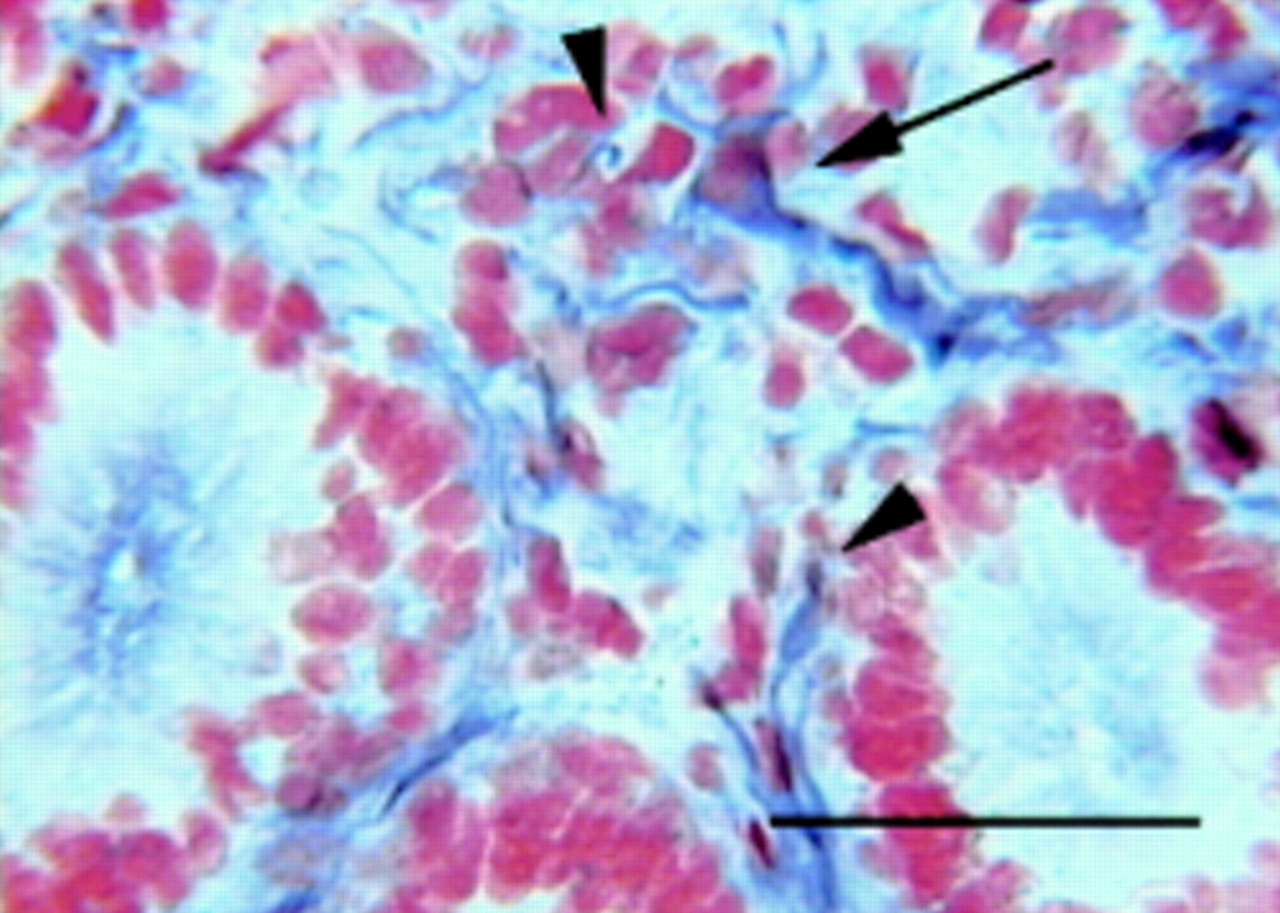
Human small intestinal mucosa. Arrowheads show apposition of the PrPc positive nerve fibres to lymphoid and epithelial cells; arrow indicates a cell showing the characteristics of an enteroglial cell. 3F4 monoclonal antibody immunohistochemistry, avidin:biotinylated enzyme complex/alkaline phosphatase with nitroblue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate as substrate. Nuclei were stained with neutral red. Scale bar represents 50 μm (originally published in Nat Med 2000; 6 :840–1).
We often observed apposition of PrPc positive nerve fibres not only to epithelial but also to lymphoid cells in the lamina propria (fig 1). These findings hint at a possible role for PrPc in the local regulation of immune and epithelial functions.
Further studies of cellular and molecular mechanisms involved in oral transmission of TSEs are essential. This may enable prevention of disease spread and development of early intervention strategies. The potential role of cellular prion protein in neuroimmunomodulation may represent a new and exciting opportunity for investigation. An understanding of the functional biology of PrPc in health is vital to future research.
Functional biology of cellular prion protein
There is considerable uncertainty about the biological function of PrPc. It has been shown recently that PrPc is copper binding and has antioxidant activity. This enzymatic property is dependent mainly on copper binding to the octarepeats region. A pathogenetic factor in prion diseases could be due to imbalances in metal catalysed reactions resulting in a decrease in antioxidant function and an increase in oxidative stress.56PrPc may also function as a signal transduction protein, as demonstrated by caveolin-1 dependent coupling of PrPc to the tyrosine kinase Fyn.57 PrPc deficient mice (Prnp0/0) have been used to elucidate the functional role of PrPc further, and a recent intriguing study shows that loss of PrPc alters the intracellular calcium homeostasis of cultured cerebellar granule cells.58 There was no evidence that this change was due to direct alteration of voltage gated calcium channels, as suggested previously.59
Iatrogenic transmission and gastroenterologists
Iatrogenic CJD was first reported after corneal implant surgery in 1974, and subsequently pituitary hormone injections were implicated. Iatrogenic CJD cases have developed after grafting cadaveric dura mater contaminated with CJD prions.60 Brain surgery itself is probably associated with an increased risk of CJD.61Currently, there is no evidence that gastrointestinal endoscopy has any potential role in transmitting PrPSc. However, in known vCJD patients a specific dedicated endoscope is used throughout the UK as otherwise the endoscope cannot be reused and destruction of the instrument is recommended. Given the long incubation period of vCJD, further information regarding the safety of endoscopy in silent carriers is required and must be a public health priority.

{kind=link}
{kind=link}
Acknowledgments
Work in the authors' laboratory is supported by a strategic grant from the Medical Research Council.
Abbreviations used in this paper
- TSE
- transmissible spongiform encephalopathy
- BSE
- bovine spongiform encephalopathy
- vCJD
- “new variant” Creutzfeldt-Jakob disease
- PrPc
- normal host encoded cellular prion protein
- PrPSc
- pathological prion protein
- LR
- laminin receptor
- CNS
- central nervous system
- FAE
- follicle associated epithelium
- FDC
- follicular dendritic cell
- ENS
- enteric nervous system
References
Footnotes
Leading articles express the views of the author and not those of the editor and editorial board.