Article Text

Statistics from Altmetric.com
The possibility that gastrin may play a role in the development of colorectal carcinoma has aroused considerable interest over the past decade. In early reports some colorectal carcinomas and colorectal carcinoma cell lines were shown to produce gastrin,1 to express gastrin receptors,2 and to respond mitogenically to exogenous gastrin.2 The most favoured explanation for these observations was an autocrine or paracrine loop in which gastrin was produced by the tumour, bound to tumour receptors, and stimulated tumour growth. However, gastrin may also have acted as an endocrine agent as hypergastrinaemia has been reported in a number of patients with colorectal carcinoma, although the source of the gastrin has not been defined.3-5 Further studies of the expression of gastrin6-8 and gastrin receptors9 ,10 in colorectal carcinomas and colorectal carcinoma cell lines, and of hypergastrinaemia in patients with colorectal carcinoma,11-15 indicated that the early positive reports were not universally correct. Recent information on the nature and source of the gastrin produced, and the discovery of novel receptors selective for non-amidated gastrins, makes it timely to reconsider the involvement of progastrin derived peptides in colorectal carcinoma.
Synthesis, storage and secretion of gastrin in colorectal carcinomas
There is now abundant evidence that most colorectal carcinomas synthesise progastrin. Gastrin mRNA has been detected by both polymerase chain reaction and northern hybridisation in colorectal carcinoma cell lines, normal human colonic mucosa and colorectal carcinomas.6 ,16-18 The gastrin mRNA is of low abundance but the major band of 0.7 kilobases is identical in sequence to antral gastrin mRNA.6 ,16 ,17 There is, however, considerable disparity in the literature on the proportion of colorectal carcinomas that contain amidated gastrin (table 1). The variable efficiency of translation and extent of postranslational processing of gastrin in peptide producing tumours20 offers an explanation for some of the contradictory reports as assays for gastrin have frequently been confined to amidated forms. The processing of gastrin from progastrin to the amidated product involves a number of steps, which include endopeptidase and carboxypeptidase mediated cleavages, and which end in conversion of glycine extended gastrin to the amidated forms (fig1).21 ,22 Using either region specific antisera or processing independent analysis,20 which quantitates all molecular forms of gastrin irrespective of the degree of processing, progastrin or progastrin derived peptides are now being detected in 80–100% of colorectal carcinomas (table 1). The presence of non-amidated gastrin peptides in colorectal carcinomas has assumed greater importance with the recent reports that gastrin-gly has a growth promoting effect in a non-transformed colonic epithelial cell line,23 as well as in a pancreatic cancer cell line24 ,25 and in fibroblasts,26 and with the description of receptors capable of binding non-amidated gastrin.23 ,24 ,26 ,27 In addition, transgenic mice expressing human progastrin in the liver had raised concentrations of plasma progastrin and a hyperplastic colonic mucosa.28 The nature of the receptor responsible for the proliferative effects of progastrin is completely unknown.
Frequency of gastrin positive colorectal carcinomas
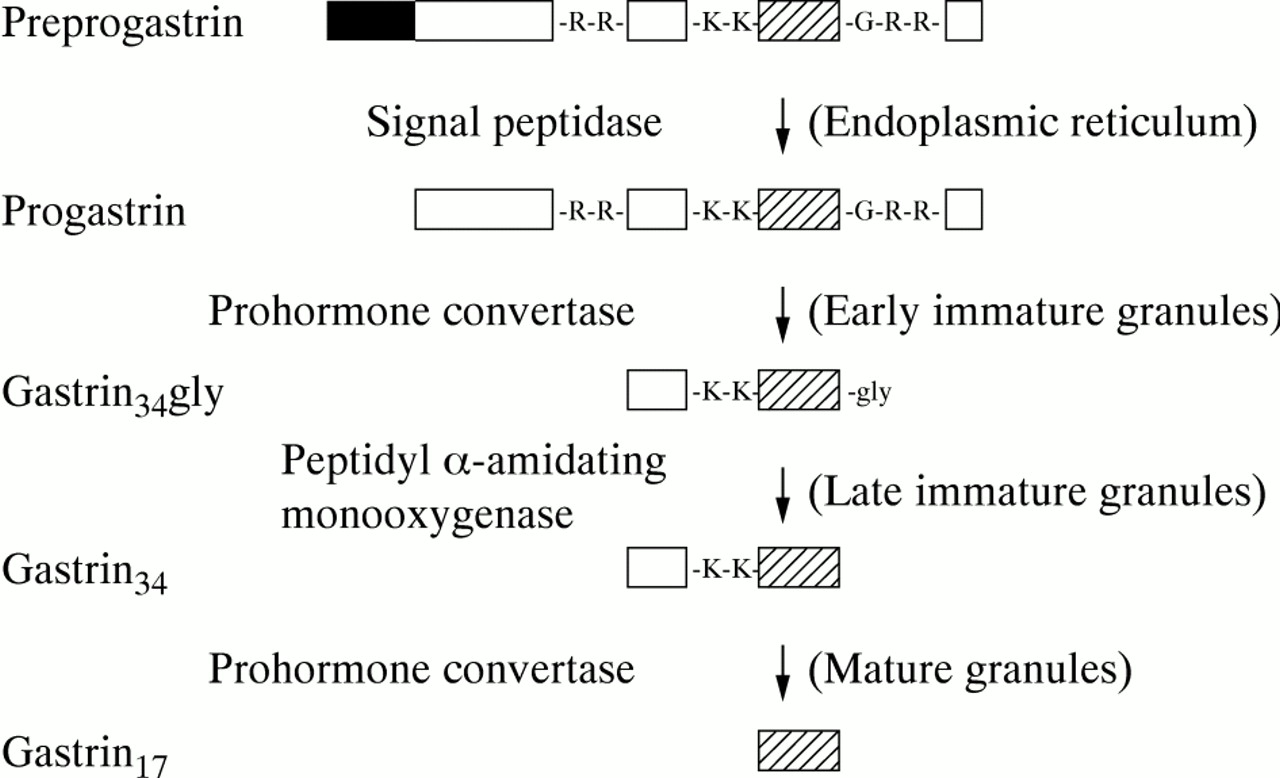
Progastrin processing. Preprogastrin is converted to gastrin34gly by sequential removal of the signal peptide (black box), and cleavage at paired arginine (R) residues by prohormone convertase(s).21 ,22 Amidation of gastrin34gly is followed by cleavage at the paired lysine (K) residues to yield gastrin17 (hatched box). Alternatively cleavage of gastrin34gly results in gastrin17gly. Note that gastrin17gly may be a distinct end product, which in rat antrum at least is not converted to gastrin17. 22 The regions of the cell in which the processing steps occur are shown in brackets.
Hypergastrinaemia in patients with colorectal carcinoma has also been controversial. Early studies were not controlled for Helicobacter pylori status (a known cause of hypergastrinaemia), had small sample sizes with the results skewed by apparent outliers, and measured only amidated gastrin.14 ,15 After controlling for these factors, we have confirmed that circulating amidated gastrin is not raised in patients with colorectal carcinoma, but have made the intriguing finding that circulating non-amidated gastrin is raised both in H pylori positive (5.2-fold) and negative (2.3-fold) patients with colorectal carcinoma.15 The initial reports of a decrease in plasma gastrin following surgical resection of the colorectal carcinoma4 ,5 have not been confirmed however.13 ,14 The low gastrin concentration in colorectal carcinomas, together with the increase in circulating partially processed gastrin, is consistent with constitutive secretion, whereby gastrin is not stored in a processed form in tumour cells, but is secreted from peptide containing vesicles by exocytosis.29An alternative explanation is that colorectal carcinomas are secreting an agent such as gastrin-releasing peptide which stimulates the secretion of antral gastrin.4
Gastrin/cholecystokinin receptors in carcinoma tissue and on cell lines
For gastrin to function as a growth promoting agent, specific receptors capable of transducing a mitogenic signal must be localised on the tumour. At least four receptors exist for gastrin and cholecystokinin (CCK).30 The classic CCK-A receptor on pancreatic acinar cells is selective for sulphated CCK8(dissociation constant (Kd) = 20 pM), whereas the gastrin/CCK-B receptor on gastric parietal cells recognises gastrin17 and both sulphated and unsulphated CCK8 with approximately equal affinity (Kd = 2–6 nM). Both A and B receptors are members of the family of receptors with seven transmembrane segments. Despite sharing 50% identity in sequence, the A and B receptors can be clearly distinguished by several selective antagonists. A low affinity gastrin17 binding site (Kd = 200 nM) has also been described on colorectal carcinoma cell lines,9 ,31 identified as a 78 kDa protein,32 and called the gastrin/CCK-C receptor.32 Surprisingly, the gastrin/CCK-C receptor is unrelated to the A and B receptors in sequence, but belongs to a family of enzymes involved in the β-oxidation of fatty acids.33The C receptor can also be distinguished from the A and B receptors by the fact that addition of a C-terminal glycine residue to gastrin17 does not affect binding.27 A novel high affinity receptor selective for glycine extended gastrin, with a different antagonist profile from the A and B receptors, has also been described on a rat pancreatic carcinoma cell line (AR4–2J),24 and on a non-transformed colonic epithelial cell line.23 A slightly different receptor, recognising both glycine extended and amidated gastrins with similar high affinity, has been reported on the mouse colon cell line CA, and on Swiss 3T3 fibroblasts.26 Definition of the relation of the novel receptors to each other, and to the gastrin/CCK-A, -B, and -C receptors, will require data on their size from cross-linking experiments and on their amino acid sequence from cloning experiments.
Controversy exists over whether most colorectal carcinomas or colorectal carcinoma cell lines express the gastrin/CCK-B receptor (summarised in table 2). Although Thompson’s group34detected high affinity gastrin binding sites on over 50% of human colorectal carcinomas, recent studies have not detected high affinity gastrin binding in resected human colorectal carcinomas,35 ,36 and mRNA encoding the gastrin/CCK-B receptor was detected in less than 20% of specimens.35 ,37 Similarly, although binding sites for the gastrin/CCK-B receptor antagonist L365,260 were detected on all four human colorectal carcinoma cell lines,38 gastrin binding sites were found on only one of 10 human colorectal carcinoma cell lines,10 and mRNA encoding the gastrin/CCK-B receptor was present in only one (LoVo) of eight colorectal carcinoma cell lines tested.37 In agreement with this observation gastrin dose dependently increased LoVo cell number39 and DNA synthesis,37 but did not affect proliferation of the other eight colorectal carcinoma cell lines tested.37 Although few studies of CCK binding in human colorectal carcinoma have been undertaken, the data available suggest that CCK-A receptor expression in such tumours is rare (table 2).36 Taken together these findings suggest that CCK-A and gastrin/CCK-B receptors are present in only a small subset of colorectal carcinomas.
Frequency of gastrin/CCK receptor positive colorectal carcinomas
Gastrin as an autocrine growth factor
Gastrin is an established growth factor for the non-antral gastric mucosa,40 but proliferative effects on the normal mucosa of the small and large intestine, or on the development of colorectal carcinoma, are controversial.30 ,41 For example, patients with hypergastrinaemia associated with the Zollinger-Ellison syndrome have an increased rate of colonic mucosal cell proliferation, but do not have an increased prevalence of colonic adenomas.42Similarly, patients with hypergastrinaemia associated with pernicious anaemia do not have an increased long term risk of colorectal carcinoma,43 although the risk may be increased in the first five years after diagnosis.44 Variations in the source and forms of gastrin may be responsible for these contrasting viewpoints as the observation that overexpression of intact progastrin in transgenic mice increased colonic mucosal proliferation suggests that biological activity is not confined to the amidated or glycine extended forms.28
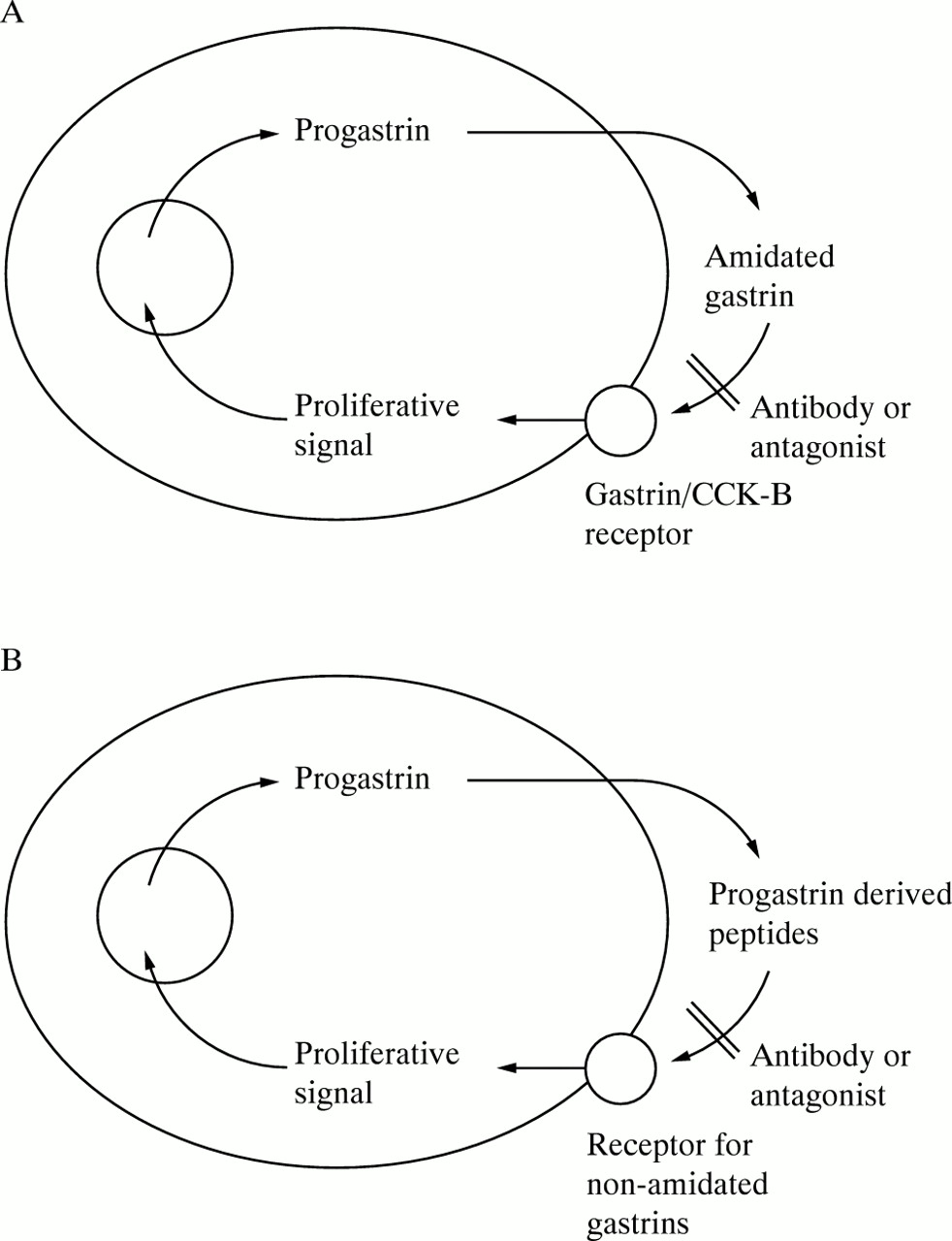
The growth of some colorectal carcinoma cell lines both in culture and as xenografts is stimulated by exogenous and endogenous hypergastrinaemia,30 ,45 and considerable interest has therefore been generated by the hypothesis that progastrin derived peptides may be acting as autocrine growth factors in colorectal carcinoma. In the autocrine model (fig 2) a cell synthesises its own growth factor, which is released into the surrounding medium. Binding of the growth factor to cell surface receptors then results in the transmission of a mitogenic signal to the cell nucleus, with a consequent increase in cell proliferation. The most convincing evidence for the involvement of progastrin derived peptides in an autocrine loop is the observation that expression of antisense gastrin mRNA reduced both growth and tumorigenicity of the colorectal carcinoma cell lines Colo 320 and HCT 116,46 and growth of the conditionally immortalised mouse colon cell line YAMC.23 In contrast, growth of the colorectal carcinoma cell line Colo 205A, which expressed negligible amounts of gastrin mRNA, was unaffected by expression of antisense gastrin mRNA.46 The antisense experiments demonstrate that progastrin derived peptides act as autocrine growth factors in colorectal carcinoma cell lines, but do not define the form of progastrin derived peptides or the nature of the receptor involved in the loop.
{kind=link}
{kind=link}
Autocrine growth loops involving progastrin derived peptides. At least two classes of autocrine growth loops involving progastrin derived peptides are possible. In both classes colorectal carcinoma cells produce and process progastrin, and the progastrin derived peptides are released into the surrounding medium. Binding of progastrin derived peptides to a specific receptor then results in an increase in cell proliferation. (A) In class I the cells secrete mature amidated gastrin, and express the gastrin/CCK-B receptor. The absence of gastrin/CCK-B receptors from most colorectal carcinoma specimens (table 2) and from colorectal carcinoma cell lines suggests that class I loops are infrequent. (B) In class II processing of progastrin is incomplete, and the cells express a surface receptor selective for non-amidated forms of gastrin, such as the receptor for glycine extended gastrins first described by Yamada and coworkers on the surface of the rat pancreatic carcinoma cell line AR4–2J.24 In principle, either loop could be blocked by expression of antisense gastrin mRNA,23 ,46 by antibodies against the appropriate progastrin derived peptides23 ,47or their receptors, or by selective receptor antagonists. Such antagonists have not yet been described for receptors for non-amidated gastrins.
The existence on colorectal carcinoma cell lines of an autocrine loop involving amidated gastrin and the gastrin/CCK-B receptor (fig 2A) does not seem to be a common phenomenon. As discussed in the last section, most colorectal carcinoma cell lines do not express gastrin/CCK-B receptors. In addition the selective gastrin/CCK receptor antagonists L364,718 and L365,260 had no effect on proliferation of colorectal carcinoma cell lines at concentrations sufficiently high to saturate CCK-A and gastrin/CCK-B receptors, respectively.48 ,49Antisera directed against the common C-terminal tetrapeptide of gastrin and CCK inhibit the proliferation of only a small proportion of colorectal carcinoma cell lines.1 ,31 ,50
The possibility of an alternative autocrine loop which utilises non-amidated gastrins (fig 2B) should also be considered. Such peptides have been demonstrated in colorectal carcinoma specimens, as discussed in the section on synthesis and secretion. In the case of the mouse colon cell line YAMC, proliferation is partially inhibited by an antibody selective for glycine extended gastrin, but not by an antibody selective for amidated gastrins.23 Furthermore, YAMC cells do not bind amidated gastrin, but do express a receptor selective for glycine extended gastrin,23 similar to the receptor first described by Yamada and coworkers on the rat pancreatic carcinoma cell line AR4–2J.24 Thus YAMC cells seem to utilise non-amidated progastrin derived peptides, and a receptor selective for glycine extended gastrin, in an autocrine loop.
The anti-proliferative effects of the non-selective gastrin/CCK receptor antagonists proglumide and benzotript on colorectal carcinoma cell lines in vitro,31 and on colorectal carcinomas grown as xenografts in nude mice51 and rats,52 seem to be mediated by a completely different mechanism as the receptor for glycine extended gastrins does not recognise either antagonist.23 In fact, a comparison of inhibitory potencies suggests that the low affinity gastrin/CCK-C receptor is the probable target as in this case binding of gastrin to the gastrin/CCK-C receptor is blocked by proglumide and benzotript, but unaffected by L364,718 and L365,260.32 Interestingly, inhibitory effects of proglumide on proliferation have also been described on fibroblasts53 ,54 and on cell lines of diverse origin,55 in agreement with the widespread tissue distribution of the gastrin/CCK-C receptor (Mantamadiotis and Baldwin, unpublished data). Clear definition of the role of the gastrin/CCK-C receptor in the autocrine effects of progastrin derived peptides will require the development of potent and selective antagonists.
Therapeutic possibilities
The presence of the autocrine gastrin loops described earlier suggests that gastrin/CCK receptor antagonists might be useful in the treatment of colorectal carcinoma. The non-selective antagonist proglumide inhibited the growth of colorectal carcinoma cell lines in vitro31 and in vivo.51 ,52 However, a clinical trial of proglumide in patients with gastric carcinoma did not reveal any benefits, perhaps because the concentrations achieved were not sufficient to saturate gastrin/CCK receptors.56 Other more potent gastrin/CCK receptor antagonists have also been shown to inhibit the growth of colorectal carcinoma cell lines in vitro,48 ,57 ,58 and of primary human colorectal carcinomas in vitro59 and in athymic rats,45but have not yet been subjected to clinical trials. The observation that most colorectal carcinomas do not express gastrin/CCK-B receptors, however, indicates that it will be unlikely that gastrin/CCK-B receptor selective antagonists will be a general treatment for colorectal carcinoma.
In contrast, antibodies directed against gastrin may be useful for treatment of colorectal carcinoma. As mentioned earlier, antibodies recognising the C-terminal amidated tetrapeptide common to both gastrin and CCK inhibit the proliferation of some,1 ,31 but not all,49 colorectal carcinoma cell lines. Conversely, proliferation of the mouse colon cell line YAMC is inhibited by antibodies recognising glycine extended, but not amidated, gastrins.23 A particularly promising development has been the demonstration that preimmunisation of rats with Gastrimmune (a conjugate of amino acids 1–9 of gastrin17 and diphtheria toxoid) reduced the in vivo growth of the rat colorectal carcinoma cell line DHDK12.47
Most colorectal carcinomas produce progastrin, but only a subset of colorectal carcinomas expresses classic gastrin/CCK-B receptors. Novel gastrin/CCK receptors capable of binding non-amidated forms of progastrin derived peptides have also been described in the past three years. High affinity antagonists selective for the non-classic gastrin/CCK receptors are required urgently, in order to test the working hypothesis that one or more of the novel receptors is the target for the autocrine proliferative effects of progastrin derived peptides. In the longer term the interaction between progastrin derived peptides and the proteins altered by the cascade of genetic mutations associated with the development of colorectal carcinoma60needs to be resolved. Demonstration of proliferative effects of progastrin derived peptides in the early stages of the adenoma–carcinoma sequence might have diagnostic and therapeutic benefits.