Article Text

Statistics from Altmetric.com
Understanding the genetic basis of “complex disease” has been heralded as one of the major challenges of the post genome era.1 However what are “complex diseases” and how will understanding the genetics of such diseases advance medical science? There has been a great deal of “hype” about the potential of the human genome mapping project.2 The three major claims are that this information will: (a) be used in diagnosis; (b) provide useful prognostic indices for disease management (including the development of individualised treatment regimens, based on the findings of both immunogenetic and pharmacogenetic studies); and (c) provide insight into the pathogenesis of these diseases. Of these three objectives the last has the greatest potential and is the least exaggerated claim. In this review I shall highlight major associations, discuss some of the practical issues that arise, and outline how current knowledge of the immunogenetic basis of chronic liver disease is beginning to inform the debate about disease pathogenesis.
AN INTRODUCTION TO COMPLEX TRAITS
Complex traits are defined as those where inheritance does not follow a Mendelian (that is, simple) pattern. This is not a new science; studies of complex traits were first performed in the mid-20th century. However, in the 21st century, the prevailing language has changed. Previously complex traits were called “polygenic” (involving more than one gene), multifactorial (depending on the interaction of the host genome and one or more environmental factors), or oligogenic (whereby individual mutations in several different genes in one or more common pathways lead to the same clinical syndrome but each patient with the disease may possess a single disease causing mutation only) (see box 1).
Box 1 Genetic diseases
Mendelian disease
-
Autosomal recessive
classical Mendelian pattern applies.
-
Autosomal dominant
classical Mendelian pattern applies.
-
Autosomal co-dominant
classical Mendelian pattern applies.
-
Sex linked
classical Mendelian pattern applies.
Complex disease
-
Oligogenic
single mutations in each case or family but several different genes make up the disease.
-
Polygenic
involves multiple genes that interact to create a permissive gene pool for disease genesis.
-
Multifactorial
as polygenic but requires environmental input for disease genesis.
The best definition of a complex trait is: where “one or more genes acting alone or in concert increase or reduce the risk of that trait”.3 This definition allows for all of the above possibilities (oligogenic, polygenic, and multifactorial), as well as non-Mendelian single gene disease, and has three important details. Firstly, genetic variations (mutations or polymorphisms (see box 2)) result in differences in the risk of disease; the disease causing mutations (DCMs) or alleles do not by themselves confer disease, they simply increase or reduce the likelihood that a trait will be expressed in a given individual. The second detail concerns the term “trait” as opposed to “disease”. Most diseases are heterogeneous syndromes and the clinical phenotypes are often extremely variable. Using the term “trait” allows for this variation in the disease phenotype and the prospect that DCMs not only determine which diseases we are more likely to develop but also may determine the severity of the clinical syndrome that follows.
Box 2 Key definitions
-
Locus—the location of a gene.
-
Allele—a genetic variation at a locus.
-
Haplotype—a series of genes inherited “en bloc” (that is, a single unit).
-
Linkage disequilibrium—product of non-random inheritance of specific alleles encoded at different gene loci whereby combinations of alleles are found together more frequently than predicted by Mendel’s law of independent segregation.
-
Mutation—a genetic variation present in less than 1% of the population.
-
Polymorphism—a mutation present in more than 1% of the population.
-
SNP—single nucleotide polymorphism. This involves exchange of one nucleotide base (ACT or G) for another.
-
λs is derived from:
the prevalence of disease in siblings/the prevalence of disease in the population
-
λs values represent the increased risk of disease to the sibling of a affected individual. Values for λs for autosomal dominant diseases may be as high as several thousand, for autosomal recessive diseases as high as several hundred but for complex disease range from 3 to 100. In autoimmune diseases, these values are usually less than 50 and greater than 5.
MEASURING THE HERITABLE COMPONENT IN COMPLEX DISEASE
Before we attempt to identify disease genes, we need to have some estimate of the heritable component of the disease so that we can determine the best and most pragmatic means of investigating it. Simple measures of heritability include: concordance in monozygotic and dizygotic twins, the degree of familial aggregation, and calculations such as sibling relative risk (λs, box 2). No such data exist for type 1 autoimmune hepatitis (AIH) and primary sclerosing cholangitis (PSC) or viral liver disease but the λs for primary biliary cirrhosis (PBC) is 10.5,4 and approximately 1 in 20 patients have an affected family member.
It is also useful at this stage to consider both the number of genes that may be involved and the size of effect anticipated. In the absence of whole genome scanning data, the best guide to this may be to compare with other diseases which have similar levels of heritability. The two most appropriate models for autoimmune liver disease are insulin dependent diabetes mellitus (IDDM, λs = 15)5 and Crohn’s disease (CD, λs = 13–36).6 Both CD and IDDM are polygenic diseases.6,7 There is significant linkage on five chromosomes (6, 12, 14, 16, and 19) in CD,6 and so far three different candidate disease gene loci on chromosomes 2, 6, and 11 have been identified in IDDM.7 Therefore, using CD and IDDM as our models, based on a λs value of 10.5 (for PBC)4 (which is lower than for CD but similar to that for IDDM), and assuming that lack of data indicates a lesser heritable component in type 1 AIH and PSC, we may expect at least 3–5, but perhaps more, disease alleles if we propose a polygenic/multifactorial model for autoimmne liver disease. It is more difficult to estimate the number of genes that influence disease outcome following viral infection.
In addition, the size of the effect varies. The risk of disease associated with known disease alleles in CD and IDDM varies from an odds ratio of 1.3 for the CTLA4 CT60 SNP in IDDM,8 to more than 20 for homozygous carriers of CARD15 mutations in CD,9 or HLA DR3/DR4 heterozygotes in IDDM.10 These differences may cloud our judgement in assessing the end use of such discoveries.
IDENTIFYING GENES IN COMPLEX DISEASE
How then can we identify genes in complex disease? There are two broad strategies: linkage and association analysis.
Linkage analysis is preferable where large collections of multiplex families are available for study. However, for many diseases, including autoimmune and viral liver diseases, large collections of multiplex families are simply not available. This problem applies to all of the less common diseases, many of those diseases where onset occurs later in life, and also to infectious diseases where transmission is horizontal (through the population) rather than vertical (through the family).
In these diseases, association analysis is the only practical option. This may be based on studies of limited pedigrees (intrafamilial association analysis—for example, the transmission disequilibrium test) or comparison of allele distribution in unrelated cases and unaffected “healthy” controls. Analysis may be based on a single candidate polymorphism, several polymorphisms in a single gene, a chromosomal region, multiple genes in a pathway, or may involve scanning the whole genome.
The advantages and disadvantages of case control association studies have been the subject of much debate,11 and I will not repeat that debate here. It is however important to note that the one major advantage of case control studies is the potential to study very large numbers. Thus these case control studies can have very high levels of statistical power, allowing identification of genes with very low risk ratios.
Despite this apparent advantage, case control studies have fallen into disrepute among geneticists because very few studies based on large numbers have been conducted. Consequently, most published studies have been woefully underpowered and many published associations have not been reproduced. However, we should not be too pessimistic; valuable lessons have been learned11 and a significant number of genetic associations, so identified, including many of those discussed herein, have been confirmed and will stand the test of time.
CANDIDATE SELECTION: WHERE TO LOOK
Apart from the statistical considerations (summarised in box 3, and addressed by Colhoun and colleagues11 and earlier reviews therein), the major issue in study design is candidate selection or where to look. Candidate genes for genetic studies are selected on the basis of: their location in the genome (if linkage data are available); prior knowledge of similar (related) diseases; knowledge of disease pathogenesis; or knowledge of the gene and its function. Aside form linkage based candidates, all of these involve a degree of speculation. One means of increasing the chance of success is to consider findings from other similar diseases or syndromes. The rationale for this is based on the principle that not all disease alleles will be disease specific. In fact, a considerable number of disease alleles will be non-specific disease promoter alleles that map to common or shared pathways. For example, disease progression in patients with liver disease is often related to the degree of fibrosis. It therefore seems reasonable to speculate that there may be a common profibrotic genotype in patients with rapidly progressing liver disease.12 Some other examples include the CTLA4 gene (below) and the cytokine gene cluster on chromosome 5q. Animal models, especially knockouts and knockins, may also be informative in selecting candidate genes to investigate.
Box 3 Statistical considerations
-
Too few patients—false negative associations, inadequate statistical power.
-
Too few controls—false negative associations, inadequate statistical power.
-
Poor matching of patients and controls—gives rise to false positive and false negative associations. Associations can arise as a result of confounding factors that differ between patients and controls rather than as a consequence of genetic differences.
-
Failure to correct for multiple testing—gives rise to false positive associations. Although correction is punitive, especially for investigators who are more thorough and have tested more possibilities, the need for correction is based on statistical consideration. The alternative to correction is to verify (replicate) in subsequent studies.
-
Failure to replicate (verify) findings of study in a second cohort—may give rise to false positive and perhaps false negative associations. This is related to need to correct for multiple testing.
-
Post hoc hypothesis development—usually involves multiple testing of disease subgroups. This is caused by the search for significant probability values to enhance publication potential, most often produces irreproducible associations based on small numbers. Such post hoc associations should be replicated.
Currently, for those diseases where linkage data are unavailable, microarray analysis and whole genome association analysis (WGAA) based on regularly spaced single nucleotide polymorphisms (SNPs) are seen as methods of choice with which to identify potential disease genes and areas of the genome for further study. Microarray analysis provides a powerful tool that may guide candidate selection, provided the tissue samples for study are adequate and sufficiently homogeneous in terms of clinical diagnosis, disease phenotype, and cell types for study. WGAA is an untested possibility with the potential to beguile as well as to inform and will require very large numbers of patients. The number of SNPs to be tested in a WGAA will be determined once extensive SNP haplotype maps of the human genome become available.
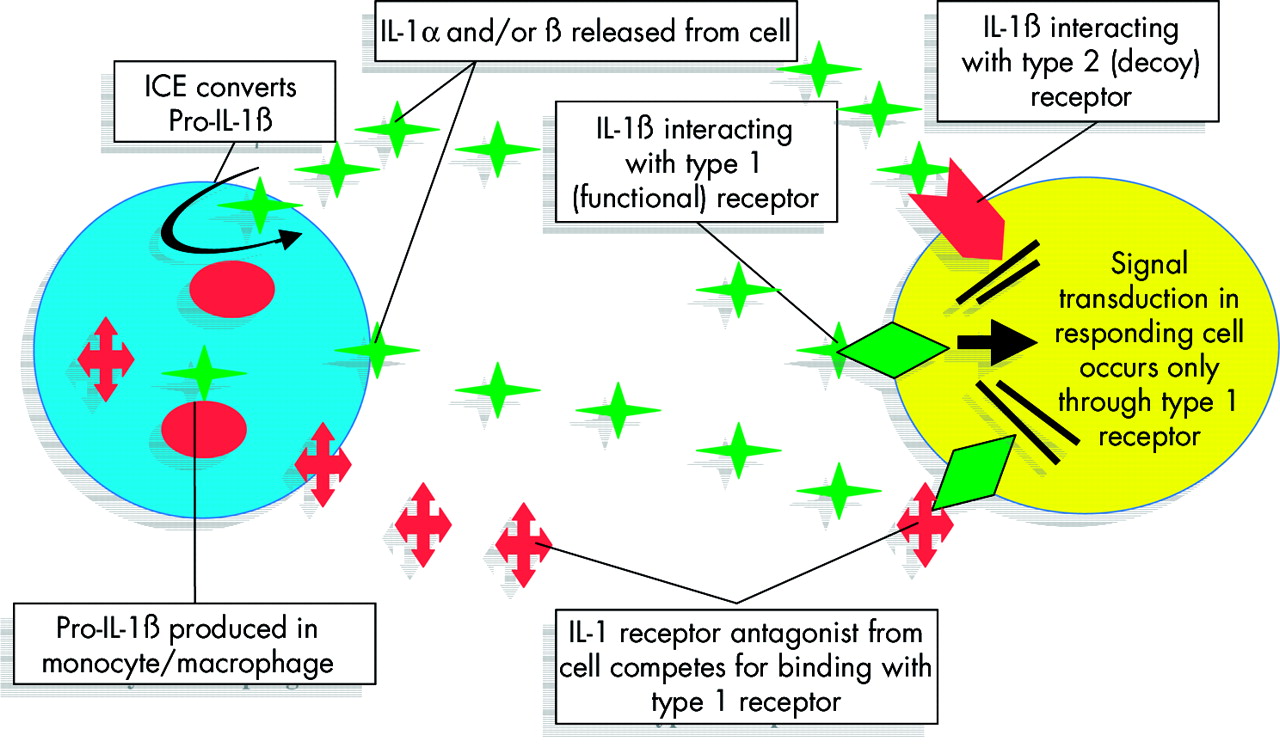
The experts advise that when selecting mutations or polymorphisms to be studied, one should be mindful of both their function and biological relevance.13 The former requirement is often difficult to satisfy because our knowledge of gene function lags far behind our knowledge of the genome sequence. Furthermore, assessing the functionality of a given polymorphism is not a straightforward process. Most genes have several polymorphic sites, many of which may impact on gene expression; SNPs are inherited as haplotypes and the potential for post-translational modification of expressed genes means that simply assessing gene transcription is not evidence of a biological effect. Even if an SNP is associated with a “functional” effect, there is no guarantee that the associated function will have a biological consequence. Most biological systems have a degree of built-in redundancy to cope with relative differences in key components. Biological pathways are complex. A good example of such a system, relevant to complex disease genetics, concerns the proinflammatory cytokine interleukin 1 (fig 1).

Interleukin 1 (IL-1) regulation: a simplified example of a complex system. The IL-1 gene family includes genes for two agonist forms of IL-1 (α and β) and a receptor antagonist (IL-1ra). In addition, there are at least two different IL-1 receptors (IL-1r1 and r2). The agonists IL-1α and IL-1β (represented by green stars) interact with the first of these receptors (IL1-r1 green diamonds) but the IL-1 receptor antagonist (red crosses) competes for binding with IL-1r1, blocking the IL-1 agonists (IL-1α or β), and the second receptor (Il-1r2, red arrow) acts as a decoy receptor, binding surplus IL-1α and IL-1β. In addition, transcribed IL-1β is produced in an inactive form within the cell (pro-IL-1β) and requires activation by an IL-1 converting enzyme (ICE, also known as caspase 1) for release into the extracellular compartment. All of the genes in this family are polymorphic. This greatly oversimplified example shows how naive the requirement for “functional status” may be when applied to a single nucleotide polymorphism (SNP) or single gene locus in isolation, representing a single element in such a tightly controlled system. In the above example, how biologically meaningful is it to consider “functionality” of a specific SNP or SNP haplotype at the IL1B locus without also considering the other members of the IL-1 family?
Candidate selection is made easier in infectious diseases where much of the pathogenesis has already been mapped out and therefore more direct questions can be addressed. A good example of this is the current work on the interferon pathway, referred to below.
INTRODUCTION TO IMMUNOGENETICS
Immunogenetics concerns that branch of genetics that deals with the genes which regulate the immune response. Immunogenetics arose in the 1960s born out of interest in the human leucocyte antigens (HLAs) and their role in transplant acceptance and rejection. The idea that the immune response is under genetic control however predates the clinical need to study HLA, and its origins can be traced back to earlier in the 20th century, with truly great scientists (Landsteiner, Gorer, Snell, Medawar, and Dausset, to name but a few). (Current and up to date nomenclature for HLA systems can be found at www.anthonynolan.org.uk). Immunogenetics was never a science that was restricted to studies of HLA. The ABO blood groups, immunoglobulin, and complement gene polymorphisms have long been included in the list. More recently, however, we have come to understand the extent of polymorphism in the human genome, and have realised almost any gene that encodes an immune active product can act as an “immune response gene.”
The immunogenes that have been studied in complex liver disease include: several members of the immunoglobulin superfamily (HLA, immunoglobulin, and T cell receptor genes); the major histocompatibility complex (MHC) encoded complement genes (C2, C4A, C4B, and factor B or Bf); a number of different cytokine genes (currently identified genetic polymorphisms in cytokine genes are summarised at www.pam.bris.ac.uk/services/GAI/cytokine4); accessory molecules (including cytotoxic T lymphocyte antigen 4 (CTLA4)); and more recently various members of the interferon pathway. Of these the associations with MHC encoded HLA genes, particularly the HLA class II DR and DQ genes, have been the most consistent (tables 1–3).
Summary of associations with human leucocyte antigen (HLA) haplotypes in autoimmune liver disease
HLA AND AUTOIMMUNE LIVER DISEASE
Studies in PBC,14 the most common of the autoimmune liver diseases, consistently report a weak but significant association with HLA DR8 (DRB1*0801 in Europeans and DRB1*0803 in Japan). The risk of disease for Europeans with the DRB1*0801 allele may be up to 8.16 times greater than for those without (table 1). However, in Europeans, this association accounts for only 15–25% of patients,14 which may indicate that the true association lies elsewhere along the chromosome, with DRB1*0801 acting as a linkage marker. These findings are not in keeping with the picture of PBC as a T cell mediated autoimmune disease. Studies of the T cell response in PBC have also failed to link DR8 alleles with presentation of key immunogenic peptides.15 Instead, the responsive T cells appear to be restricted by the HLA DRB4*0101 allele, which encodes DRw53, a very common HLA antigen found in more than 50% of most populations but which is not associated with susceptibility to the disease. This is not informative in terms of the pathogenesis of PBC, other than in a negative sense, suggesting that some thought should be given to the role of non-classical MHC and other immunogenes in PBC, and that immunological investigations should not be restricted to the activities of T cells.
The strongest HLA associations reported in autoimmune liver disease are those for PSC and type 1 AIH (table 1). However, despite apparent similarities between these two diseases, the reported associations appear to map to different gene loci within the MHC. Thus in PSC, MHC encoded susceptibility appears to be involving either a combination of DR, DQ, and MHC class I chain-like (MIC) alleles or perhaps MIC alone,16,17 whereas in type 1 AIH, MHC encoded susceptibility appears to be related very closely to specific DRB1 alleles that carry lysine or arginine at position 71 (fig 2).18

Lysine-71 and susceptibility to type 1 autoimmune hepatitis (AIH). Lysine-71 and arginine-71 versus alanine-71 basis of major histocompatibility complex (MHC) encoded susceptibility and resistance to type 1 AIH. DRB1 alleles, amino acid motifs (DRβ-67 to DRβ-72), and amino acids at position-71 of the DRβ polypeptide. The key molecular differences between human leucocyte antigen alleles that encode susceptibility to type 1 AIH (represented in yellow) and those that encode resistance (represented in blue) are shown. Within the six amino acid motif represented by the single letter code LLEQKR (positions 67–72), the critical amino acid appears to be that found at position 71—namely, lysine (K) or arginine (R) on susceptibility alleles and alanine (A) on resistance alleles. The three amino acids vary in terms of polarity and overall charge, as illustrated. Both lysine and arginine have two positively charged amino groups and a single (negatively charged) carboxyl group while alanine has only a single amino group and carboxyl group. This may be the basis of MHC encoded genetic susceptibility to type 1 AIH.
This difference has important implications in terms of disease pathogenesis. In PSC, the potential involvement of the MIC genes indicates a more prominent role for the innate immune response.19 MICα molecules appear to be expressed exclusively on gastrointestinal and thymic epithelia and are seen in non-diseased liver.20 MICα molecules may be induced by stress and heat shock and have been identified as a ligand for γδ T cells, natural killer (NK) (CD56+) cells and T cells expressing the NKG2D activatory receptor.21 Engagement of this receptor activates γδ and NK cell effector functions. Although the functional significance of MICA*008 homozygosity remains to be determined, the “normal” liver has a large resident population of γδ T cells, NK, and natural (N)T cells.22 Increased numbers of γδ and NK cells have been documented in PSC livers.23,24 If PSC arises as a result of infection, this may provide the catalyst for heat shock induction of MICα on biliary epithelium leading to activation of intrahepatic γδ and NK cells with subsequent cytokine secretion and cytolytic effector functions. The MICA*008 allele carries a short tandem repeat sequence with a premature stop codon. It has been suggested that this may lead to aberrant or unstable expression of the MICα protein and that in individuals homozygous for MICA*008, this may promote persistent immune activation leading to autoimmunity or failed activation with the consequence of persistent infection with an increased likelihood of collateral damage and ultimately the recognition of self (auto) antigens.
That PSC genetics may be more complex also fits with the characteristics of this disease. PSC does not fulfil the classical criteria for an autoimmune disease (organ specific autoantibodies, female preponderance, and responsiveness to corticosteroid therapy). The common overlap with inflammatory bowel disease, mostly ulcerative colitis, and high incidence of malignancy are both potential indications that this is a heterogeneous syndrome with several interrelated pathologies. Hence a more complex genetic picture may be anticipated.
In contrast with PSC, type 1 AIH is a classical autoimmune disease. The DRB1 association in type 1 AIH points to the MHC class II antigen presenting pathway and events in T cell activation as the keys to understanding disease initiation (fig 3). This fits well with the characteristics of type 1 AIH as a predominantly T cell mediated disease. However, the lysine-71 model (fig 2) does not tell us much about the autoantigen in type 1 AIH and is not universal. Indeed, an alternative model based on valine/glycine dimorphism at position-86 of the DRβ polypeptide has been proposed, which better represents the key HLA associations in patients from Argentina and Brazil,25 but does not fit the European/North American data.18 In a study of Japanese patients with type 1 AIH,26 all were found to have DRB1 alleles which encode histidine at position-13. These three different models may suggest that different genetic associations have arisen in different populations and they may be related to differences in endemic viruses. It is interesting to note that in South America where hepatitis A virus is endemic, persistent infection is associated with carriage of the HLA DRB1*1301 allele,27 the same allele that characterises the majority of children who develop type 1 AIH in that population.25 Thus these HLA associations may be the molecular footprints of the prevailing environmental triggers that precipitate type 1 AIH. These triggers will vary from population to population. This hypothesis is pure speculation as a common viral trigger has yet to be defined for type 1 AIH but recent studies indicate that an infectious agent may be responsible for the genesis of PBC.28
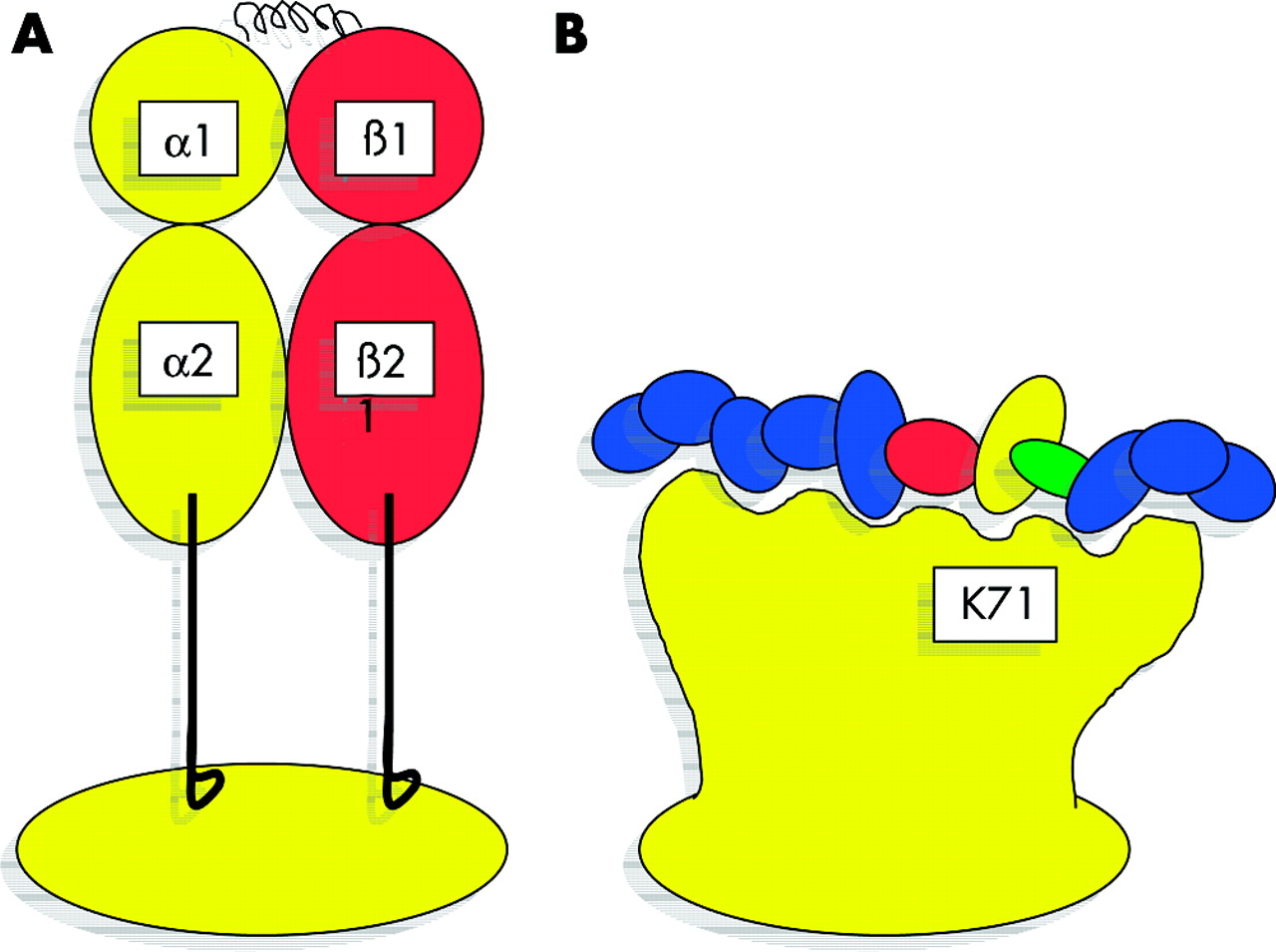
Representation of human leucocyte antigen class II molecules and relationship between lysine at position 71 of the DRβ polypeptide and the bound peptide. The expressed major histocompatibility complex (MHC) class II molecule is a heterodimer of two polypeptide chains, each with two Ig domains. (A) The structure is folded to create a platform supporting a single antigen binding cleft into which short antigenic peptides are bound. The majority of genetic polymorphism results in amino acid substitutions in and around the antigen binding cleft. Substituting amino acids at critical sites may profoundly affect physical interaction between the MHC molecule and the antigenic peptide, and may favour binding of peptides with specific physical properties. In particular, the MHC binding cleft has pockets along its length where there is interaction with the side chains of the antigenic peptide. Molecular changes will also influence the orientation of the bound peptide in the cleft and therefore may influence the dynamics of the interaction with the T cell receptor. In this diagram (B), the peptide, illustrated by the coloured ovals, is shorter than that usually found in an MHC class II molecule. You are asked to imagine that possession of lysine at position 71 (K71) favours binding of a peptide which has the red-yellow-green motif (residues 4–6 counting in from the right hand side of the picture). Perhaps peptides with this red-yellow-green motif will be more efficiently bound by MHC molecules that possess lysine at position 71 than those with alanine-71. Were this red-yellow-green motif to be an essential component of the autoantigen for type 1 autoimmune hepatitis, more efficient interaction between lysine 71 may be an important factor in promoting disease in those with lysine-71 bearing alleles, whereas alanine, with less efficient binding, may have a protective effect.
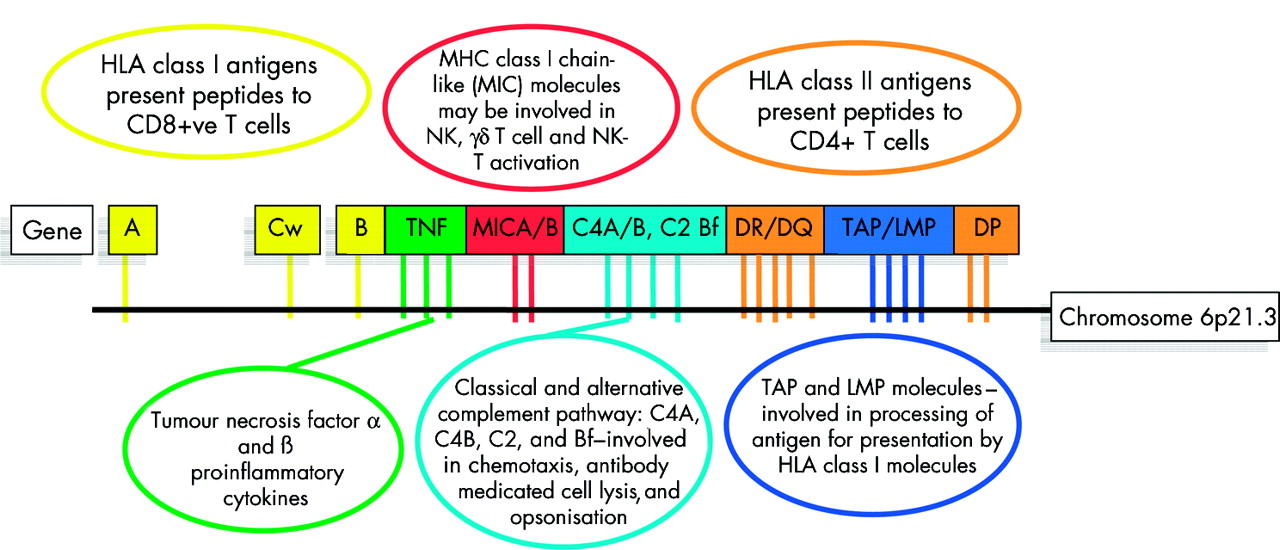
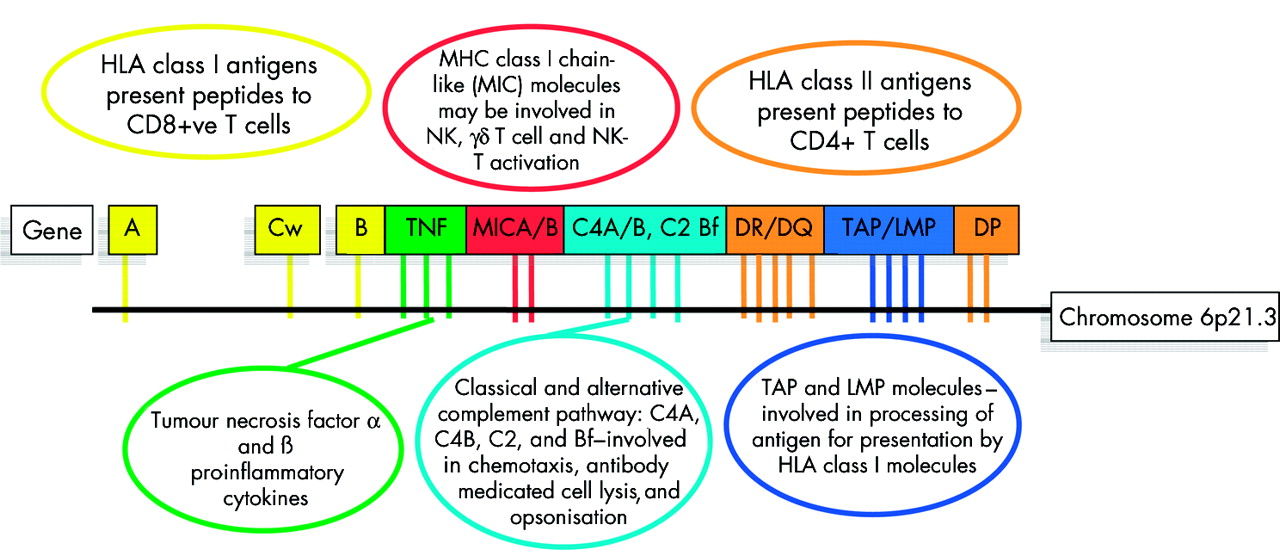
Lysine-71 and other models for type 1 AIH do not fully explain the disease. In European and North American patients, those with DRB1*0301 or the extended haplotype (short hand HLA 8.1) have quite different clinical characteristics to those with the DRB1*0401 haplotype.18 Both haplotypes carry DRB1 alleles that encode lysine-71; therefore, other genes within the MHC (or closely linked genes) must also be active and may modify the clinical phenotype. Traditional candidates include the MHC encoded complement and tumour necrosis factor genes which all map to the class III region (fig 4). However, there are many new recently characterised genes in this region including MICA and MICB (MHC class I chain related A and B genes) that may also serve as candidates.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Key genetic components of the major histocompatibility complex (MHC) and their main functions in immunobiolgoy. The human MHC encodes nearly 200 genes in 4 Mb of DNA. The key components of the MHC include the classical transplantation antigens HLA A, B, Cw, DR, DQ, and DP (yellow and light brown) but the region also includes key complement genes (light blue), tumour necrosis factor genes (TNF) (green), and other genes with critical function in both innate and adaptive immunity. All of the genes illustrated are polymorphic and thus the extended MHC haplotypes may carry multiple disease promoting or disease resistance alleles.
HLA AND VIRAL HEPATITIS
The relationship between host HLA and viral hepatitis has been explored (table 2). In most cases the risk of infection is not itself genetically determined but host genes do play a role in determining outcome following exposure. Thus individuals who have the DRB1*1301 allele are more prone to persistent hepatitis A virus infection,27 those with DRB1*1302 are more prone to persistent hepatitis B virus (HBV) infection,29 and those with DQB1*0301 haplotypes are more likely to have self-limiting hepatitis C virus (HCV) infection.30,31
Summary of key human leucocyte antigen (HLA) haplotypes associated with acute (self-limiting versus protracted (chronic)) infection in viral hepatitis
In the European population, data on HCV are particularly pertinent. The majority of those infected by this virus become chronic carriers and the virus is present in a significant proportion of the European population. We may expect that individuals with DQB1*0301 would have a more vigorous immune response to HCV peptides, and indeed they do.32 In addition, studies have shown that these patients have a greater response to a range of HCV peptides and are more likely to have a multispecific response as opposed to a monospecific response when challenged with different HCV derived peptides in vitro. (Cramp M, et al, unpublished data 2003). This may be important in combating the mechanisms by which the virus evades the host immune response. Studies have shown that to eliminate the virus, a multispecific immune response is required. Interestingly, work based on the structure of the DQB1*0301 molecule has failed to identify a single DQB1*0301 specific HCV epitope (Klenerman P, personal communication, June 2003). Perhaps it is the promiscuity of the DQ heterodimer involving the DQB1*0301 allele which favours binding of many different viral epitopes and thereby promotes viral clearance in those with this allele.
It is interesting to note that the key HLA haplotypes in viral infection are also implicated in AIH and PSC (tables 1, 2). Although there is no absolute correlation between haplotypes that promote viral persistence and those which promote autoimmune disease, there is at least some overlap; DRB1*0301 in some studies of HCV33 and DRB1*1301 (above) for example.16,17,25,27,29 In contrast, DRB1*1501 which protects from type 1 AIH in Europeans18 promotes HCV persistence in most studies, whereas DRB1*0701 which may protect from PSC17 may also promote HCV persistence. This provides a basis for speculation about the overlap between autoimmunity and viral disease and it is hoped that as we accumulate more data on all of these diseases these relationships may become clearer.
NON-HLA IMMUNOGENETICS
Thus far very little is known of the role of the other antigen presenting genes in either autoimmune or viral liver disease. Early studies on Ig and T cell receptor (TCR) genes in type 1 AIH and more recent studies of the MHC encoded TAP genes in PBC and HCV have not been replicated.
Although antigen presentation is critical, there are many other stages in the immune response that may be governed by host genetic variation. Critical stages in the immune response include the immediate aftermath of MHC-peptide-TCR interaction where signalling by accessory molecules determines the course of events. One of these accessory molecules, the cytotoxic lymphocyte antigen 4 molecule (CTLA-4) is especially interesting. Switching from immune activation to immune memory occurs through upregulation of CTLA-4 on CD25+ T cells. There are numerous SNPs in the CTLA4 gene on chromosome 2q33 and a number of these have been implicated in susceptibility to autoimmune disease.8,34 Two of these have received particular attention, A+49G34 and more recently CT60.8 Preliminary studies of the A+49G SNP have identified potential associations with both PBC35 and type 1 AIH36 while work on PSC is ongoing (Broome U, personal communication, September 2003). The association with PBC has not been replicated in all populations studied (Baragiotta A, et al, unpublished data, 2003), which may reflect a closer linkage with the functional SNP CT608 or the relatively small number of patients studied in the original publication.35 The absence of an association with CTLA4 in PBC may once again bring into question the role of the T cell in this disease.
As T cell immune responses predominate in immunity to HBV and HCV, we may also expect CTLA4 to have a potential role in determining the outcome following viral infection. However, studies in our centre suggest that there are no major associations with the A+49G SNP and either acute self-limiting HCV infection or persistent HBV infection (Agarwal K et al, unpublished data 2003). There is however a single report of an association with response to interferon/ribavarin in HCV but until this has been confirmed it is of little use clinically or otherwise.37
The immune response is orchestrated, at least in part, by the cytokine network. Cytokines have multiple functions and act through specific cell surface receptors. The cytokine network is extremely complex and interactive. Genetically determined relative differences in cytokine production may decide whether a cell is activated or not, and by activating different cells whether activation leads to a predominantly T helper (Th)1 or Th2 immune response.
Early studies of the tumour necrosis factor-α gene (TNFA) stimulated interest in investigating other cytokine genes in disease, starting with the proinflammatory cytokines, interleukins (IL)-1 and -6, interferon γ, then spreading to the immunoregulatory cytokines IL-4 and IL-10. For the most part these studies examined preselected single SNPs or occasionally multiple SNPs in a single target gene. Although only a limited number of cytokine genes have being investigated so far, apart from some preliminary data on the IL-1 gene family in PBC38 and the IL-10 promoter polymorphisms in alcoholic liver disease39 and HCV,40 these studies have mostly drawn a blank, and even those studies which have reported positive associations38–40 are controversial. The reason for the lack of success and/or consistency in the majority of investigations lies with the complexity of the cytokine network, the limited scope, and the relatively small numbers included in most of the studies. More recent work on cytokine gene receptors promises to be more inclusive and may herald some potentially important findings in relation to both HCV and HBV.41 In future, studies based on large collections of well characterised patients employing high throughput genotyping, to explore whole pathways and networks, will radically alter our perception of case control association studies, and should eliminate many of the discrepancies which dominate the published literature, and perhaps restore the reputation of the case control approach to disease genetics.
WHAT THE STORY OF CARD15 IN CROHN’S DISEASE CAN TEACH US ABOUT THE GENETICS OF OTHER SIMILAR COMPLEX DISEASES
Much can be learned about potential disease genes by looking at studies of other diseases. Just as other diseases may indicate which genes to investigate, they may also provide clues about problems and also help us to understand what to expect from genetic analysis in complex diseases. The CARD15 story in CD is an excellent example and shows us how genetics can inform pathogenesis.
CARD15 encodes the human protein nucleotide binding oligomerisation domain 2 (NOD2). NOD2 is one of a family of pattern recognition receptors or proteins. NOD2 mediates recognition of bacterial peptidoglycans through its leucine rich repeat (LRR) domains and signals through two caspase recruitment domains (CARDs). Signalling induces nuclear factor κB, caspase activation, and secretion of proinflammatory cytokines IL-1β and TNF-α. In CD, it appears that mutations in the LRRs of the CARD15 gene lead to absent or decreased signalling and defective host responses to bacterial infection.
A number of important lessons can be learned from the CARD15 story. Firstly, CARD15 was identified in CD following whole genome scanning and exhaustive rounds of fine mapping.42,43 The genetic association in CD is based on three SNPs which all have the same effect on NOD2 function. Overall, each individual SNP has only a small impact on disease risk but the cumulative effect of all three SNPs on the CD population generates an odds ratio of 3 for heterozygotes and up to 23.4 for homozygotes.9 Taken individually, differences in the case versus control frequencies of all three SNPs are quite small, and because of this any case control study based on less than 200 patients and controls would have failed to identify the CARD15 association. This illustrates the importance of larger numbers in case control studies. Secondly, there are marked differences in the distribution of the three SNPs between populations. Thus the most common SNP in the UK population is SNP89 whereas the most common SNP in the New York Ashkenazi Jewish population is SNP12.44 If we were to investigate SNP12 in isolation in the UK using a case control approach, we would most likely miss the association altogether as SNP12 is found in only 3.4% of UK CD and 0.6% of UK controls.9 To reach statistical significance (even at the 5% level), this difference requires a sample of approximately 500 cases and controls and few investigations in liver disease have been based on more than 100 or 200 patients. Not only does this reinforce the first lesson above but it also illustrates the importance of analysing all three SNPs and not restricting the analysis to single SNP in the hope of finding an association. Thirdly, in addition to the three common mutations (SNPs 8, 12, and 13), a significant proportion of CD families (up to 25%) have rare mutations in the CARD15 gene45 which could not be detected in a simple case control study. This highlights the major strategic weakness of case control studies; the inability to detect associations based on individual mutations.
Bearing in mind the relationship between inflammatory bowel disease and PSC, it may at first seem reasonable to investigate CARD15 in this disease. However, the most common form of inflammatory bowel disease to occur in PSC is ulcerative colitis, which is not associated with CARD15 mutations, and CD is a relatively rare overlap in PSC. Mutations in the NOD domain of the CARD15 gene are however associated with another disease characterised by granulomatous inflammation, Blau’s syndrome. In Blau’s syndrome there is uncontrolled signalling from the CARD domains. As granuloma is common in PBC, CARD15 may seem a worthy candidate to investigate in PBC.
THE ROLE OF INTERFERON GENES IN DETERMINING OUTCOME FOLLOWING HCV INFECTION
Recent work in viral hepatitis aimed at identifying non-MHC determinants of disease outcome, employing high throughput saturation analysis of key genes and their SNPs, has identified three potentially important associations.41 The associated mutations are in the MXA, PKR, and OAS-1 genes, all of which are involved in the response to interferon.41 Of these, the association with the MXA-88 SNP has been reported previously46 while the other two are novel. MXA is an antiviral protein induced by interferon. OAS-1 encodes the enzyme oligo-adenylate synthetase which catalyses formation of RNAseL, which breaks down viral RNA. PKR is an RNA dependent protein kinase that halts viral replication. Clearly this type of approach is the way forward in complex disease—using high throughput technologies to screen polymorphisms in major components of biologically relevant pathways as opposed to selecting single SNPs and single genes for study—but the real key to success and consistency will be to look at much larger numbers, thousands rather than hundreds.
Summary
-
Understanding the genetic basis of complex disease is a major challenge that will offer new approaches to disease diagnosis and patient management but mostly it will help us to understand disease pathogenesis.
-
Immunogenetics is concerned with interindividual variation in the genes that control the immune response. Most studies to date have been concerned with the human MHC, but an increasing number of investigators are now focussing on other immunogenes that affect both innate and acquired immunity to self and foreign antigens.
-
Immunogenetic studies in liver disease have detailed MHC encoded susceptibility to a number of different autoimmune and viral liver diseases (tables 1–3). These studies highlight important differences and similarities between diseases, and also implicate different pathways in disease genesis.
-
In type 1 autoimmune hepatitis, a model has been created whereby HLA DRB1 alleles encoding specific amino acid motifs may best explain MHC encoded disease susceptibility and resistance. This model is in keeping with type 1 AIH as a T cell mediated disease. Furthermore, there is clear genetic evidence for the possibility of overlap between susceptibility to viral hepatitis and susceptibility to type 1 AIH in some populations. Perhaps HLA associations tell us more about susceptibility to potential infectious triggers than they do about the self-antigens.
-
In contrast, recent studies of primary sclerosing cholangitis may indicate that other pathways are operating. In particular, the association with the MHC encoded MICA alleles may indicate a role for non-classical T cell or NK cells.
-
The MHC appears to have a lesser role in PBC where the key associations are linked with disease progression rather than susceptibility per se.
-
There are clear indications that the host MHC genes influence the outcome following exposure to hepatitis A, B, and C viruses. These observations may have implications for vaccine design, as well as helping us understand the mechanisms involved in viral clearance.
-
Overall studies of non-MHC genes have been disappointing. The problem lies with study design. Many lessons have been learned and new and interesting associations are beginning to emerge, in particular in relation to interferon therapy in HCV infection.
-
Much more work is required but we are now beginning to see some of the fruits of our labours, if not in terms of understanding disease pathogenesis then at least in our efforts to focus the questions.
-
By applying new technologies, such as expression array analysis and high throughput genotyping, rapid advances will be made in understanding the genetics of complex liver disease. Although genetics will provide only one part of the jigsaw in understanding disease pathogenesis, it is anticipated that this knowledge will help us to find and put together the remaining pieces.
Summary of confirmed associations
DISCUSSION
To what extent have we started to realise the promise that immunogenetics will inform the debate on disease pathogenesis in complex liver disease? Clearly there is still much to be done. The overall picture is mixed; negative and controversial claims do little to illuminate disease pathways. We cannot dismiss a protein or pathway simply because there is no genetic association. There are some high points. Lysine-71 has the potential to be informative in type 1 AIH,18 and DQB1*030130,32 may help us to understand self-limiting HCV infection and this latter information may ultimately be useful in vaccine design. Furthermore, the recent MXA, PKR, and OAS-1 data in HCV41,46 may help to make sense of the variable response of HCV patients to interferon therapy, and may ultimately be useful in informing the decision about who to treat, and what treatment options to apply in different cases. These data may also help us to understand why so many infected individuals become chronic carriers of the HCV virus.
The MICA data on PSC provides a strong alternative hypothesis to simply investigating T cell phenomena in that disease. The strong protective effect of MICA*002 and the finding of a very significant association with MICA*008 homozygotes but not heterozygotes suggests a mechanism by which faulty MIC expression in those with MICA*008 may lead to increased immune activity.19 Some effort should now be given to understanding this relationship and the role of MIC in innate immunity and this disease. In addition, it is important to explore the genetic basis for the high rate of overlap with ulcerative colitis and the high rate of malignancy in PSC. Identifying clinical subsets within PSC that may enable us to make more sense of the disease pathology is one way to unravel this complex syndrome.
In PBC, HLA has a limited effect,14 which may indicate that antigen presentation through the MHC class II pathway is not of paramount importance in disease pathogenesis. In other words, the T cell immune response may be an important effector mechanism in disease genesis but (perhaps in contrast to type 1 AIH) this is not simply a matter of erroneous recognition of self-antigens which are preferentially presented by DR8 (DRB1 alleles expressing lysine-71 in type 1 AIH). The key to understanding PBC is probably not the T cell itself; we have identified the antigen, we must now concentrate on understanding how and why immune tolerance to pyruvate dehydrogenase complex E2 (antigen) is compromised. The genetic association with interleukin 138 may provide some insight but clearly there are many other candidates in this disease.
Do any of the observations above surprise us and what do these findings indicate for future studies? That most of the non-MHC associations in autoimmune liver diseases are weak and controversial should not surprise us. These are relatively rare disorders; large collections are difficult to assemble and studies in other diseases indicate that non-MHC genes may have only small effects in autoimmune disease.8 Furthermore, it is difficult to predict genetic effects in viral disease where there are many confounding variables that influence the outcome.
That studies of the MHC are more consistent is also not surprising. The genetic associations with HLA are stronger in most autoimmune diseases than those for non-MHC genes. The HLA polymorphisms are functional and biologically relevant but perhaps there is an additional reason why these associations are so strong. Closely linked with the HLA genes are several other immune response genes which encode key components in both the innate as well as the acquired immune response. Such is the degree of linkage disequilibrium within the MHC that some whole haplotypes stretching up to 4 Mb can be predicted from the identity of one or two alleles—for example, most Europeans who have HLA B8 and DRB1*0301 will have the extended 8.1 haplotype which includes MICA*008, TNFA*2, C4AQ0, as well as DQA1*0501 and DQB1*0201. These alleles encode the unstable MICα chain, the high producer TNF-α phenotype, null or non-expressed forms of complement C4, and the HLA DQ2 molecule, respectively. Although there are many extended haplotypes in the human genome, there are very few regions that display this degree of linkage disequilibrium. As a consequence of this evolutionary conservation, HLA haplotypes associated with disease may each carry several disease promoting alleles. Thus the consistency of the HLA association studies is explained, as stronger associations require fewer patients.
In future studies we need to plan for genes with low risk ratios, perhaps 1.5 or lower, in some but not all diseases. This means studying large numbers which may require collaboration across national and international boundaries, but it is imperative that we do not sacrifice quality for the sake of collaboration. The emphasis must be on well characterised patient series, not simply on the total numbers collected.
CONCLUSION
The scientific community now have the information and tools to tackle complex disease genetics. The major advances in genetics that characterised the last decade will allow us to apply reverse logic to our endeavours whereby we use genes to identify pathways for analysis rather than pathways leading us to genes. Mass screening of SNPs in biological pathways will illuminate key elements in disease pathogenesis, many of them involved in more than one disease. The challenges we face are: (a) not repeating the mistakes of the past11,13; (b) producing large collections of well defined patients; and (c) making sense of the information that molecular genetics reveals.