Article Text

Statistics from Altmetric.com
Coeliac disease is caused by T cell sensitisation of the intestine to cereal prolamins, which results in a range of mucosal abnormalities that may lead to malabsorption.1 The population prevalence in western countries is ∼1 in 200.2–4 Evidence for an inherited predisposition to coeliac disease comes from studies of first degree relatives of patients and studies of twins.5,6 A strong association is seen between coeliac disease and the HLA-DQ (α1*05, β1*02) heterodimer (DQ2) which is present in approximately 95% of patients,7–9 compared with 20-30% of healthy subjects.10,11 The difference in concordance rates between monozygotic twins and HLA identical sibs (80-100% v 25%) implicates non-HLA genes in the genetic predisposition to coeliac disease.11 The overall relative risk in sibs is at least 20 and is therefore four-fold higher than that attributable to HLA alone under model of inheritance.7 Genome linkage searches carried out on Irish,12 Italian,13, and UK14 coeliac disease families have identified a number of potential sites for the location of non-HLA linked genes. The putative candidate loci detected in the three studies are, however, largely inconsistent and the findings have not been replicated in other populations.15–19 Here we report the results of a genome screen of 24 multiplex families with coeliac disease and discuss the findings of this study in relation to previously published analyses.
METHODS AND RESULTS
Twenty-four families with two or more members affected with coeliac disease were recruited for this study (fig 1). Nine of these families (Nos 1, 3, 4, 6, 30, 32, 39, 43, and 44) have been used in a previous study of candidate regions.16 All the families were of northern European ancestry. Twelve of the families were recruited from the UK, nine from Sweden, two from Switzerland, and one from The Netherlands. All affected family members had symptomatic disease and were diagnosed according to the revised criteria formulated by the European Society for Paediatric Gastroenterology, Hepatology, and Nutrition (ESPGHAN).20 Families consisted of 88 affected subjects, 36 male and 52 female, giving a male to female ratio of 1:1.5. The age at diagnosis ranged from 9 months to 74 years with a mean of 25 years. Blood samples and clinical data were all collected with informed consent and ethical review board approval. All family members were typed for the HLA-DQ A1*0501 and B1*0201 alleles by PCR-SSP.21 Ninety-nine percent of affected subjects in the families studied had HLA class II genotypes compatible with possession of the DQA1*05, B1*02 alleles.

Pedigree structures of the families studied.
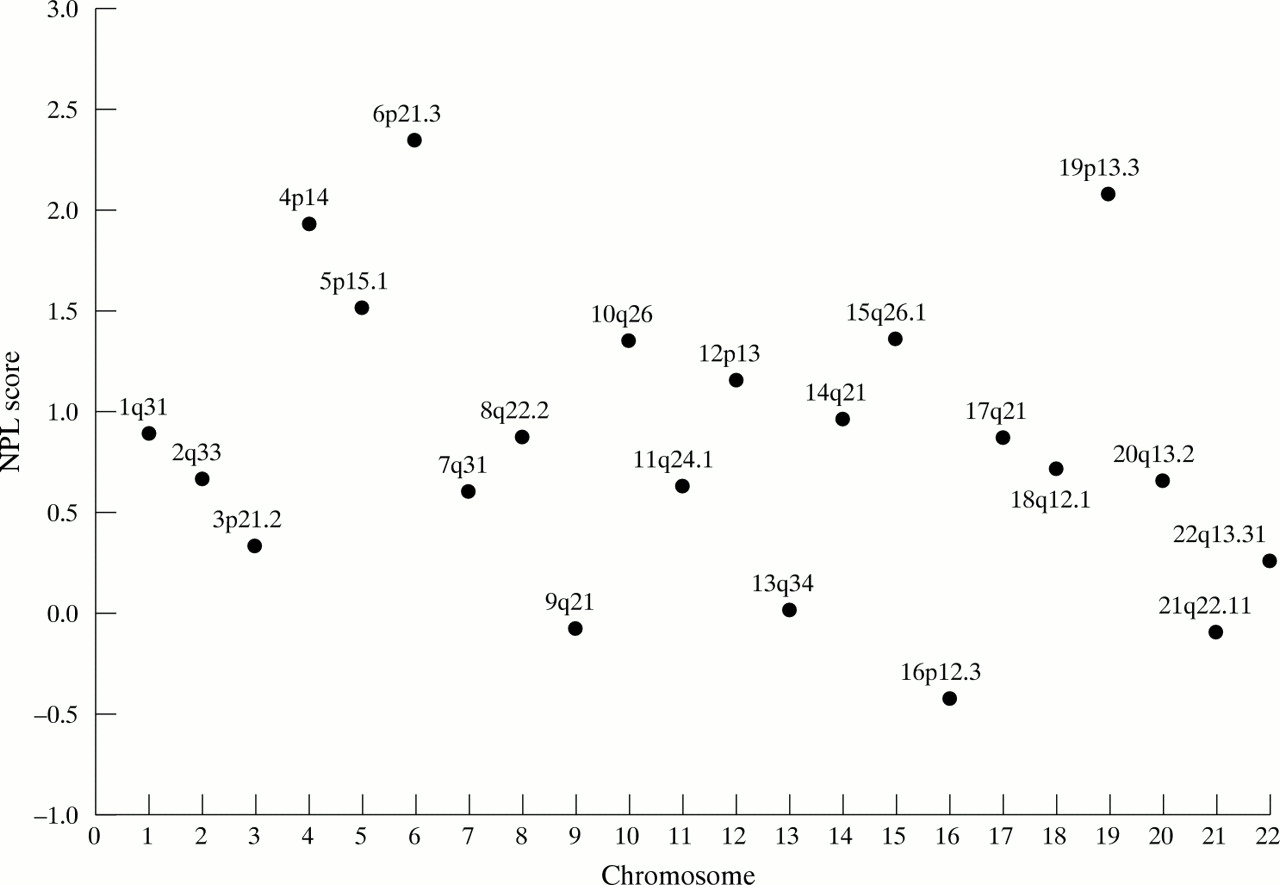
Fluorescent genotyping16 was performed at 217 microsatellite markers covering every autosome at an average distance of 16.4 cM. Genetic linkage was assessed by determining multipoint NPL (non-parametric linkage) scores using the GENEHUNTER-PLUS program.22 Multipoint NPL scores are model independent and effectively measure the extent to which marker allele sharing by descent between affected subjects is greater than expected under random segregation. Fig 2 shows the distribution of NPL scores by chromosome. The strongest region of linkage was observed at chromosome 6p21.3, at the site of HLA (nominal p value 0.01). As expected there was evidence of excess sharing at both HLA and close flanking markers and this underlies the 6p linkage. There was no evidence of linkage to any region telomeric to HLA. Outside chromosome 6p21 the strongest linkage was obtained at chromosome 4p14 (maximal at D4S405, nominal p value 0.03) and at chromosome 19p13.3 (maximal between D19S424 and D19S894, nominal p value 0.02).
{kind=link}
{kind=link}
NPL scores by chromosome.
DISCUSSION
Three genome wide linkage searches of coeliac disease have been published,12–14 but no regions of significant linkage have been reported consistently. The first of these was reported by Zhong et al.12 Linkage of coeliac disease to five chromosome regions outside HLA was detected: 6p23 (telomeric to HLA), 7q31.3, 11p11, 15q26, and 22cen. In addition to these regions, there was also some evidence of linkage to chromosome 19p13.3 (p=0.05) as seen in our study. The second study was reported by Greco et al.13 In addition to HLA, there was some evidence for linkage to 5qter and 11qter, but no evidence to support the notion that there is a locus on chromosome 6p distinct from HLA as proposed by Zhong et al.12 A follow up study of candidate regions reported by Greco et al19 failed to confirm linkage to chromosome 11qter, but gave some support for linkage to chromosome 5q (maximum MLS 2.9). We were unable to find any evidence of linkage to chromosome 5q region in our dataset. The genome wide study recently reported by King et al14 was only based on analysis of 16 UK multiplex coeliac disease families. Two regions, chromosomes 10q23.1 and 16q23.3, provided evidence of linkage in multipoint analyses using five markers (nominal p values of 0.006). There was limited evidence for linkage to HLA in the study (lod score <1.2).
In addition to a failure to confirm linkage to chromosome 6p23, neither our study nor that of Greco et al13 and King et al14 found evidence for linkage to chromosome 11p11, as originally proposed by Zhong et al.12 The proposition of an additional chromosome 6p locus made by Zhong et al12 was based on typing 30 of 45 affected sib pairs at D6S259, and flanking markers within 1 cM showed no significant evidence for linkage. In our study, there was no evidence for linkage to this region. Similarly, there is no support for linkage of coeliac disease to a locus on chromosome 6 telomeric to HLA in the studies reported by Greco et al13 and Brett et al.15
Although we found no significant evidence for linkage to chromosomes 5qter or 11qter as initially suggested by Greco et al,13 we cannot preclude these loci as sites of a non-HLA linked locus. We did, however, find some evidence for linkage at chromosome 19p13.3, raising the possibility that this region may define an additional locus.
While coeliac disease is oligogenic, the simple HLA association coupled with the fact that the environmental trigger is identical in all subjects theoretically makes genetic analysis of the trait more amenable to dissection than other complex disorders, such as non-insulin dependent diabetes mellitus or asthma. As expected, our study confirms HLA as a risk factor for coeliac disease. Two other regions showed evidence for comparable linkage, chromosomes 19p13.3 and 4p14. However, neither attains the level of statistical significance desired for genome wide linkage searches and hence require confirmation.
The genetic analysis of coeliac disease potentially has many advantages over other complex traits, both from the perspective of the magnitude of the genetic trait and the ease of defining affected status. However, the studies reported to date suggest that identifying the non-HLA linked component may not be straightforward. It is possible that the discrepancies in findings between the four studies are because of differences in the contribution of genetic factors to coeliac disease in the different populations analysed. Alternatively and specifically related to the plethora of significant findings reported by Zhong et al,12 this might reflect in part the structure of the families studied. Specifically, 31 of the ASPs belonged to three families. The inclusion of large sibships in which no typing information is available may well have led to artificially inflated support for specific regions.
If disease susceptibility is the result of more than one gene, the power to detect linkage depends on the relative contribution to the overall familial risk made by each locus and how the different loci interact. The power of this and previously published studies12–14 to detect linkage and the effect of varying heterogeneity between zero and 75% was assessed by non-parametric means using the program ALLEGRO.23 In the absence of a definitive model for the mode of inheritance of coeliac disease, genotypes were simulated under models chosen on the basis that the sib relative risk conferred by the disease gene was ∼3.3. This follows from the assumptions that the overall disease prevalence is 0.005 and that the HLA linked and unlinked genes interact multiplicatively.7 The models were a common recessive disease gene with frequency 0.2, a rare recessive disease gene with frequency 0.05, a common dominant disease gene with frequency 0.05, and a rare dominant disease gene with frequency 0.003. Even under the assumption of homogeneity, the power of each of the studies to show significant linkage over all models is marginal, especially in the study reported by Zhong et al,12 which has a maximum power of 56% to detect linkage under any of the models. The study by King et al14 is consistently less powered than the present one, and this reflects the small sample size used. In all studies, the power to detect linkage falls markedly in the presence of heterogeneity, with a power of less than 20% to detect linkage under 50% heterogeneity, under all models. This suggests that current genome wide linkage searches of coeliac disease are underpowered. If coeliac disease is heterogeneous, subcategorisation of disease by clinical phenotype or HLA haplotype might provide larger familial risks and hence increased power. Furthermore, the use of isolated groups such as the Finnish or Sardinian populations with limited genetic heterogeneity can be advantageous because of a founder effect. The optimal linkage design for the detection of a non-HLA predisposition gene for coeliac disease is not as simple as for a rare high penetrance gene. Aside from our study and that of King et al,14 the two previous genome wide searches12,13 have been based on affected sibs, excluding four of the 15 families in the study reported by Zhong et al.12 Families comprising three or more cases are likely to be more powerful for linkage than affected sib pairs or other two case families. However, increasing the number of affected subjects in a family may not necessarily represent an efficient strategy, depending on the genetic model. On the basis of segregation studies, homozygosity at an HLA unlinked locus is a perquisite for developing coeliac disease.24–26 Hence, there is a possibility that in any large family two copies of a mutation may be segregating if the population frequency of the disease allele is high. Practically, the composition of families used in linkage analysis will always depend on what can efficiently be collected. It is clear that further studies, ideally commensurate with the detection of linkage under heterogeneity, are required to support or refute the possible location of putative non-HLA linked genes.
Acknowledgments
Sanjay Popat is in receipt of a Fellowship from the Institute of Cancer Research-Royal Marsden Hospital Trust and Stephen Bevan a Fellowship from the Coeliac Society. We would like to thank all members of the families who participated in this study.